Lernergebnisse
- Identifizieren Sie häufige Fehler, die einen abnormalen Karyotyp erzeugen können
- Identifizieren Sie Syndrome, die sich aus einer signifikanten Änderung der Chromosomenzahl ergeben
Von allen Chromosomenstörungen sind Anomalien in der Chromosomenzahl am offensichtlichsten aus einem Karyogramm erkennbar. Störungen der Chromosomenzahl umfassen die Duplikation oder den Verlust ganzer Chromosomen sowie Änderungen der Anzahl kompletter Chromosomensätze. Sie werden durch Nichtdisjunktion verursacht, die auftritt, wenn Paare homologer Chromosomen oder Schwesterchromatiden sich während der Meiose nicht trennen. Falsch ausgerichtete oder unvollständige Synapsen oder eine Funktionsstörung des Spindelapparats, die die Chromosomenmigration erleichtert, können zu einer Nichtdisjunktion führen. Das Risiko einer Nicht-Disjunktion steigt mit dem Alter der Eltern.
Nondisjunktion kann entweder während der Meiose I oder II mit unterschiedlichen Ergebnissen auftreten (Abbildung 1). Wenn sich homologe Chromosomen während der Meiose I nicht trennen, Das Ergebnis sind zwei Gameten, denen dieses bestimmte Chromosom fehlt, und zwei Gameten mit zwei Kopien des Chromosoms. Wenn sich Schwesterchromatiden während der Meiose II nicht trennen, ist das Ergebnis ein Gamet, dem dieses Chromosom fehlt, zwei normale Gameten mit einer Kopie des Chromosoms und ein Gamet mit zwei Kopien des Chromosoms.
Übungsfrage
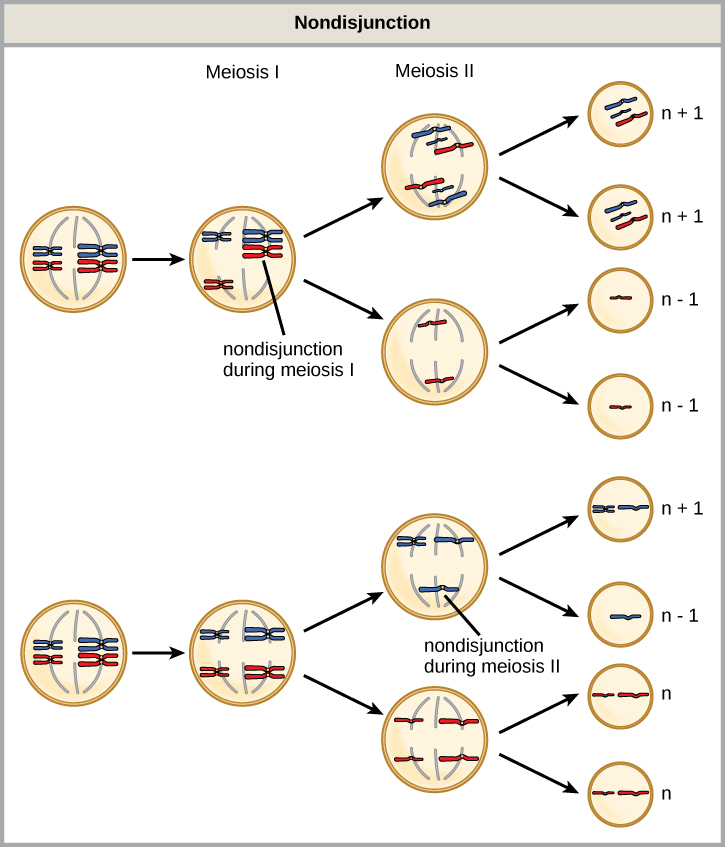
Abbildung 1. Nichtdisjunktion tritt auf, wenn homologe Chromosomen oder Schwesterchromatiden sich während der Meiose nicht trennen, was zu einer abnormalen Chromosomenzahl führt. Eine Nichtdisjunktion kann während der Meiose I oder Meiose II auftreten.
Welche der folgenden Aussagen über nondisjunction ist wahr?
- Nondisjunktion führt nur zu Gameten mit n + 1– oder n-1-Chromosomen.
- Während der Meiose II auftretende Nicht-Disjunktion führt zu 50 Prozent normalen Gameten.
- Nicht-Disjunktion während der Meiose I führt zu 50 Prozent normalen Gameten.
- Nondisjunction führt immer zu vier verschiedenen Arten von Gameten.
Aneuploidie

Abbildung 2. Die Inzidenz eines Fötus mit Trisomie 21 nimmt mit dem Alter der Mutter dramatisch zu.
Ein Individuum mit der entsprechenden Anzahl von Chromosomen für seine Spezies wird euploid genannt; Beim Menschen entspricht die Euploidie 22 Autosomen-Paaren und einem Paar Geschlechtschromosomen. Ein Individuum mit einem Fehler in der Chromosomenzahl wird als aneuploid beschrieben, ein Begriff, der Monosomie (Verlust eines Chromosoms) oder Trisomie (Gewinn eines fremden Chromosoms) einschließt. Monosomische menschliche Zygoten, denen eine Kopie eines Autosoms fehlt, entwickeln sich ausnahmslos nicht zur Geburt, weil ihnen essentielle Gene fehlen. Dies unterstreicht die Bedeutung der „Gendosierung“ beim Menschen. Die meisten autosomalen Trisomien entwickeln sich auch nicht bis zur Geburt; Duplikationen einiger der kleineren Chromosomen (13, 15, 18, 21 oder 22) können jedoch zu Nachkommen führen, die mehrere Wochen bis viele Jahre überleben. Trisomische Individuen leiden an einer anderen Art von genetischem Ungleichgewicht: einem Überschuss an Gendosis. Personen mit einem zusätzlichen Chromosom können eine Fülle der von diesem Chromosom kodierten Genprodukte synthetisieren. Diese zusätzliche Dosis (150 Prozent) spezifischer Gene kann zu einer Reihe funktioneller Herausforderungen führen und schließt häufig die Entwicklung aus. Die häufigste Trisomie bei lebensfähigen Geburten ist die des Chromosoms 21, was dem Down-Syndrom entspricht. Personen mit dieser Erbkrankheit sind durch Kleinwuchs und verkümmerte Ziffern, Gesichtsunterschiede, die einen breiten Schädel und eine große Zunge umfassen, und signifikante Entwicklungsverzögerungen gekennzeichnet. Die Inzidenz des Down-Syndroms korreliert mit dem Alter der Mutter; Ältere Frauen werden häufiger mit Feten schwanger, die den Trisomie-21-Genotyp tragen (Abbildung 2).
Polyploidie

Abbildung 3. Wie bei vielen polyploiden Pflanzen ist diese triploide orange Taglilie (Hemerocallis fulva) besonders groß und robust und wächst Blüten mit der dreifachen Anzahl von Blütenblättern ihrer diploiden Gegenstücke. (Bildnachweis: Steve Karg)
Ein Individuum mit mehr als der richtigen Anzahl von Chromosomensätzen (zwei für diploide Arten) wird polyploid genannt. Zum Beispiel würde die Befruchtung eines abnormalen diploiden Eies mit einem normalen haploiden Sperma eine triploide Zygote ergeben. Polyploide Tiere sind extrem selten, mit nur wenigen Beispielen unter den Plattwürmern, Krebstieren, Amphibien, Fischen und Eidechsen. Polyploide Tiere sind steril, weil die Meiose nicht normal ablaufen kann und stattdessen meist aneuploide Tochterzellen produziert, die keine lebensfähigen Zygoten ergeben können. Selten können sich polyploide Tiere asexuell durch Haplodiploidie vermehren, bei der sich ein unbefruchtetes Ei mitotisch teilt, um Nachkommen zu zeugen. Im Gegensatz dazu ist Polyploidie im Pflanzenreich sehr verbreitet, und polyploide Pflanzen sind tendenziell größer und robuster als Euploiden ihrer Art (Abbildung 3).
Geschlechtschromosomen-Nichtdisjunktion beim Menschen
Menschen zeigen dramatische schädliche Wirkungen mit autosomalen Trisomien und Monosomien. Daher mag es kontraintuitiv erscheinen, dass menschliche Frauen und Männer normal funktionieren können, obwohl sie unterschiedliche Anzahlen des X-Chromosoms tragen. Anstatt eines Gewinns oder Verlusts von Autosomen sind Variationen in der Anzahl der Geschlechtschromosomen mit relativ milden Effekten verbunden. Zum Teil geschieht dies aufgrund eines molekularen Prozesses namens X-Inaktivierung. Früh in der Entwicklung, wenn weibliche Säugetierembryonen aus nur wenigen tausend Zellen bestehen (relativ zu Billionen beim Neugeborenen), wird ein X-Chromosom in jeder Zelle inaktiviert, indem es sich fest zu einer ruhenden (ruhenden) Struktur verdichtet, die als Barr-Körper bezeichnet wird. Die Wahrscheinlichkeit, dass ein X-Chromosom (maternal oder väterlicherseits abgeleitet) in jeder Zelle inaktiviert wird, ist zufällig, aber sobald die Inaktivierung erfolgt, haben alle Zellen, die von dieser Zelle abgeleitet sind, dasselbe inaktive X-Chromosom oder denselben Barr-Körper. Durch diesen Prozess kompensieren Frauen ihre doppelte genetische Dosis von X-Chromosom.

Abbildung 4. Bei Katzen befindet sich das Gen für die Fellfarbe auf dem X-Chromosom. Bei der Embryonalentwicklung weiblicher Katzen wird eines der beiden X-Chromosomen in jeder Zelle zufällig inaktiviert, was zu einem Schildpattmuster führt, wenn die Katze zwei verschiedene Allele für die Fellfarbe hat. Männliche Katzen, die nur ein X-Chromosom haben, weisen niemals eine Schildpattfarbe auf. (Bildnachweis: Michael Bodega)
Bei sogenannten „Schildpatt“ -Katzen wird die embryonale X-Inaktivierung als Farbvariation beobachtet (Abbildung 4). Frauen, die heterozygot für ein X-gebundenes Fellfarbgen sind, exprimieren eine von zwei verschiedenen Fellfarben über verschiedene Regionen ihres Körpers, entsprechend dem X-Chromosom, das im embryonalen Zellvorläufer dieser Region inaktiviert ist.
Eine Person, die eine abnormale Anzahl von X-Chromosomen trägt, inaktiviert alle bis auf ein X-Chromosom in jeder ihrer Zellen. Selbst inaktivierte X-Chromosomen exprimieren jedoch weiterhin einige Gene, und X-Chromosomen müssen für die ordnungsgemäße Reifung der weiblichen Eierstöcke reaktiviert werden. Infolgedessen sind X-Chromosomenanomalien typischerweise mit leichten psychischen und physischen Defekten sowie Sterilität verbunden. Wenn das X-Chromosom ganz fehlt, entwickelt sich das Individuum nicht in utero.
Mehrere Fehler in der Geschlechtschromosomenzahl wurden charakterisiert. Individuen mit drei X-Chromosomen, Triplo-X genannt, sind phänotypisch weiblich, zeigen jedoch Entwicklungsverzögerungen und verminderte Fruchtbarkeit. Der XXY-Genotyp, der einer Art Klinefelter-Syndrom entspricht, entspricht phänotypisch männlichen Individuen mit kleinen Hoden, vergrößerten Brüsten und reduzierter Körperbehaarung. Es gibt komplexere Arten des Klinefelter-Syndroms, bei denen das Individuum bis zu fünf X-Chromosomen hat. Bei allen Arten wird jedes X-Chromosom außer einem inaktiviert, um die überschüssige genetische Dosis auszugleichen. Dies kann als mehrere Barr-Körper in jedem Zellkern gesehen werden. Turner-Syndrom, charakterisiert als X0-Genotyp (d. H., nur ein einziges Geschlechtschromosom), entspricht einem phänotypisch weiblichen Individuum mit kleiner Statur, vernetzter Haut im Nackenbereich, Hör- und Herzstörungen sowie Sterilität.
Duplikationen und Deletionen
Zusätzlich zum Verlust oder Gewinn eines gesamten Chromosoms kann ein Chromosomensegment dupliziert oder verloren gehen. Duplikationen und Deletionen produzieren oft Nachkommen, die überleben, aber körperliche und geistige Anomalien aufweisen. Duplizierte Chromosomensegmente können mit vorhandenen Chromosomen verschmelzen oder im Kern frei sein. Cri-du-chat (aus dem Französischen für „Schrei der Katze“) ist ein Syndrom, das mit Anomalien des Nervensystems und identifizierbaren körperlichen Merkmalen verbunden ist, die sich aus einer Deletion des größten Teils von 5p (dem kleinen Arm von Chromosom 5) ergeben (Abbildung 5). Säuglinge mit diesem Genotyp geben einen charakteristischen hohen Schrei ab, auf dem der Name der Störung basiert.

Abbildung 5. Diese Person mit Cri-du-Chat-Syndrom wird im Alter von zwei, vier, neun und 12 Jahren gezeigt. (kredit: Paola Cerruti Mainardi)
Probieren Sie es aus
Beitragen!
Diese Seite verbessernmehr erfahren