- Einleitung
- Materialien und Methoden
- Patienten und Proben
- Genetische Analyse
- Statistische Analyse
- Ethikerklärung
- Ergebnisse
- Klinische Merkmale von Patienten mit VHL-Krankheit
- Gesamtüberleben und CHB-spezifisches Überleben bei Patienten mit VHL-Krankheit
- Univariate und multivariate Cox-Regressionsanalysen für das Gesamtüberleben
- Auswirkungen einer unterschiedlichen Organbeteiligung auf das CHB-spezifische Überleben
- Die Auswirkung der RCC-Größe auf das Überleben der Patienten
- Erklärung zur Datenverfügbarkeit
- Ethikerklärung
- Autorenbeiträge
- Förderung
- Interessenkonflikt
- Danksagung
- Ergänzungsmaterial
- Abkürzungen
Einleitung
Die Von-Hippel-Lindau (VHL) -Krankheit (MIM 193300) ist ein autosomal dominantes Tumorsyndrom, das durch Keimbahnmutationen im Tumorsuppressorgen VHL verursacht wird, das sich auf 3p25 befindet. Die Inzidenz beträgt ~ 1 von 36.000-40.000 Lebendgeburten und hat eine hohe Penetranz (1-3). Gutartige und bösartige Tumoren, einschließlich Hämangioblastom des Zentralnervensystems (CHB), retinales Hämangioblastom (RA), Nierenzellkarzinom (RCC), Phäochromozytom (PHEO), Pankreasläsionen, einschließlich zystischer Pankreasläsionen, und neuroendokrine Pankreastumoren (PCL / PNET), endolymphatischer Sacktumor (ELST) und papilläre Zystadenome des Nebenhodens oder des breiten Ligaments sind die Hauptmanifestationen dieser Krankheit (4-11). Sowohl intra- als auch interfamiliäre phänotypische Heterogenität kann bei Patienten mit VHL-Krankheit beobachtet werden (12, 13).
Viele Studien zeigten, dass das Überleben von Patienten mit VHL-Krankheit schlechter war als in der Allgemeinbevölkerung. Eine Studie aus dem Jahr 1990 zeigte, dass das mediane Überleben 49 Jahre betrug (14). Eine kürzlich durchgeführte Studie berichtete, dass die geschätzte Lebenserwartung für männliche und weibliche Patienten 67 bzw. 60 Jahre betrug (15). Im Jahr 2012 berichtete eine Studie, dass die mittlere Lebenserwartung für Patienten mit VHL-Krankheit 52 Jahre betrug (16). Zu den zuvor berichteten Risikofaktoren für das Überleben von Patienten mit VHL-Krankheit gehörten ein frühes Geburtsjahr, weniger Überwachungsbesuche, weibliches Geschlecht, positive Familienanamnese, früher Ausbruch der Krankheit, verkürzender Mutationstyp und RCC größer als 3 cm (15, 17, 18). Allerdings hat keine frühere Forschung die Auswirkungen der Beteiligung jedes Organs in einer großen Patientengruppe untersucht.
Die Auswirkungen der Beteiligung verschiedener Organe auf das Überleben wurden bei anderen systemischen Erkrankungen untersucht. Beispielsweise weist eine Uterusbeteiligung bei Patienten mit diffusem großzelligem B-Zell-Lymphom auf ein schlechteres Überleben hin als bei Patienten mit Beteiligung anderer Organe (19). Yuda et al. berichtet, dass Perikarderguss und Beteiligung mehrerer Organe Prädiktoren für die Mortalität bei Patienten mit systemischer Leichtkettenamyloidose waren (20). Obwohl einige frühere Studien Genotyp-Phänotyp-Korrelationen bei der VHL-Krankheit festgestellt haben: Patienten mit Missense-Mutationen entwickeln häufiger ein Phäochromozytom, und Patienten mit verkürzten Mutationen entwickeln häufiger RCC und CHB (21-23). Die bestehenden Genotyp-Phänotyp-Korrelationen können andere komplexe Phänotypen nicht erklären. Daher ist es wichtig, die Prognose der VHL-Erkrankung aus der Perspektive der Organbeteiligung zu untersuchen (23, 24).
In dieser Studie zielten wir darauf ab, das Gesamtüberleben und das CHB-spezifische Überleben in einer großen Patientengruppe mit VHL-Krankheit zu untersuchen, die asymptomatische Mutationsträger umfasst, und den Einfluss von CHB, RCC, RA, PCL / PNET und PHEO zu untersuchen, um die genetische Beratung und die klinischen Behandlungsstrategien für Patienten mit VHL-Krankheit zu verbessern.
Materialien und Methoden
Patienten und Proben
Wir haben alle Patienteninformationen rekrutiert, die in der Datenbank der Patienten mit VHL-Krankheit im Ersten Krankenhaus der Universität Peking gespeichert sind. Die Diagnose wurde gestellt, wenn festgestellt wurde, dass der Patient eine Keimbahn-VHL-Genmutation trägt oder die zuvor beschriebenen klinischen Kriterien erfüllt (25, 26). In jeder Familie wurde mindestens ein Patient durch einen VHL-Mutationstest diagnostiziert, mit Ausnahme derjenigen, die den Test ablehnten. Insgesamt wurden 376 Patienten aus 134 Familien diagnostiziert. Klinische Daten einschließlich Geschlecht, Familienanamnese, Mutationstyp, Erkrankungsalter jedes Organs und Todesursache wurden durch Überprüfung der Krankenakten oder Befragung von Familienmitgliedern erhalten. Vierzig Patienten wurden aufgrund unklarer Daten von der Studie ausgeschlossen. Insgesamt 336 Patienten aus 125 Familien wurden schließlich in diese Kohorte aufgenommen, in der 298 der Patienten mindestens eine VHL-krankheitsbedingte Läsion aufwiesen und 38 Patienten asymptomatische Mutationsträger waren.
Wir haben den Überlebensstatus der 336 Patienten von der Geburt bis zum Tod oder bis zum Ende der Nachsorge im Juni 2017 aufgezeichnet. Die mediane Nachbeobachtungszeit betrug 37 Jahre / Person (Bereich 1-75 Jahre) mit insgesamt 12.966 Personenjahren.
Genetische Analyse
Ein QIAamp DNA Blood Mini Kit (QIAGEN, Deutschland) wurde verwendet, um genomische DNA aus peripherem Blut verdächtiger Patienten zu extrahieren. Drei kodierende Exons und ihre flankierenden intronischen Regionen wurden mittels PCR unter Verwendung der zuvor beschriebenen Primer amplifiziert (27). Missense-Mutationen, Spleißmutationen und kleine Indels wurden durch direkte Sequenzierung nachgewiesen. Ein multiplexligationsabhängiger Sondenamplifikationskit (MLPA, P016-C2, MRC-Holland, Amsterdam) wurde verwendet, um große Deletionen nachzuweisen. Große Exon-Deletionen wurden durch quantitative Echtzeit-PCR mit den von Ebenazer et al. (28). Die Patienten wurden nach den zuvor beschriebenen Kriterien in eine Missense-Mutationsgruppe (n = 162) und eine trunkierende Mutationsgruppe (n = 174) eingeteilt (18).
Statistische Analyse
Patientendemografie und grundlegende klinische Daten wurden mit deskriptiven Statistiken analysiert. Das einsetzende Alter von Läsionen in verschiedenen Organen wurde unter Verwendung des Kruskal-Wallis-Tests verglichen; paarweise Vergleiche wurden durchgeführt, und der P-Wert wurde nach Bonferroni-Anpassung bestimmt. Das Erkrankungsalter in der ZNS-Gruppe und der Abdominalgruppe wurde unter Verwendung des Mann-Whitney-U-Tests verglichen.
Da die Nachsorge von Geburt an durchgeführt wurde, haben Patienten vor und nach Ausbruch der Krankheit unterschiedliche Überlebensrisiken. Diese Situation entspricht nicht den Anforderungen des proportionalen Gefahrenmodells, wodurch die Cox-Regressionsanalyse ungeeignet ist. Stattdessen wurde die Beteiligung jedes Organs im Cox-Regressionsmodell als zeitabhängige Kovariate behandelt. Univariate und multivariate Cox-Regressionsanalysen wurden verwendet, um die Auswirkungen von CHB, RCC, RA, PCL / PNET und PHEO auf das Gesamtüberleben zu bewerten. Wir verwendeten dann univariate und multivariate Cox-Regressionsanalysen, um die Wirkung von RCC, RA, PCL / PNET und PHEO auf das CHB-spezifische Überleben zu bestimmen, da CHB die häufigste Todesursache bei Patienten mit VHL-Krankheit ist.
Die statistische Analyse wurde mit SPSS 22.0 und der Software GraphPad Prism (Version 6) durchgeführt. P < 0.05 wurde als statistisch signifikant angesehen. Für paarweise Vergleiche wurde Bonferronis Anpassung durchgeführt.
Ethikerklärung
Diese Studie wurde vom Institutional Review Board des Peking University First Hospital (Peking, China) genehmigt und von allen Patienten schriftlich genehmigt Zustimmung eingeholt.
Ergebnisse
Klinische Merkmale von Patienten mit VHL-Krankheit
CHB war die häufigste Läsion in unserer Studie, und mehr als die Hälfte der Patienten hatte CHB-Läsionen (62,2%). PCL/PNET war die zweithäufigste Läsion (46,4%). Fast zwei Drittel der verstorbenen Patienten starben an KHK (66,2%). Die Prävalenz von KHK und RCC entsprach in dieser Studie im Allgemeinen den Ergebnissen früherer Studien (ergänzende Tabelle 1). Das mediane Alter betrug ≤ 30 Jahre zu Beginn von CHB und RA und > 30 Jahre zu Beginn von RCC, PHEO und PCL/ PNET (Tabelle 1). Wir kombinierten CHB und RA in einer Gruppe des Zentralnervensystems (ZNS) und RCC, PHEO und PCL / PNET in einer Abdominalgruppe. Die Verteilung des Erkrankungsalters in den beiden Gruppen ist in Abbildung 1 dargestellt. Das mittlere Alter bei Auftreten von Tumoren in der Abdominal- und ZNS-Gruppe betrug 34 bzw. 29 Jahre. Der Mann-Whitney-U-Test zeigte, dass das Erkrankungsalter in der ZNS-Gruppe signifikant jünger war als in der Abdominalgruppe (P < 0,001).
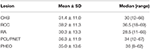
Tabelle 1. Alter zu Beginn der VHL-bedingten Läsionen.
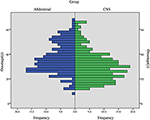
Abbildung 1. Die Häufigkeit des Auftretens des Alters in der ZNS-Gruppe (einschließlich CHB & RA-Patienten) und in der Abdominalgruppe (einschließlich RCC, PHEO & PCL / PNET-Patienten).
Gesamtüberleben und CHB-spezifisches Überleben bei Patienten mit VHL-Krankheit
Das Gesamtüberleben und das CHB-spezifische Überleben sind in den Abbildungen 2A, B dargestellt. Die mediane Überlebenszeit für Patienten mit VHL-Krankheit betrug 66 Jahre. Zusammen machten CHB und RCC 95,6% aller Todesfälle aus. Fast zwei Drittel der Patienten starben an KHK-bedingten Erkrankungen und mehr als ein Drittel der Patienten starb an RCC. Unter 209 Patienten mit CHB starben 45 Patienten an CHB, 10 Patienten an RCC. Unter 138 Patienten mit RCC starben 16 Patienten an RCC, 4 Patienten an CHB. Unter 86 Patienten mit beiden Läsionen trugen CHB und RCC gleichermaßen zu verstorbenen Patienten bei, beide verursachen 8 Todesfälle. CHB war die häufigste Todesursache als RCC in unserer Studie (Tabelle 2).
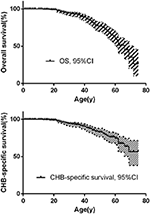
Abbildung 2. (A) Das Gesamtüberleben von VHL-Patienten. (B) CHB-spezifisches Überleben von VHL-Patienten.
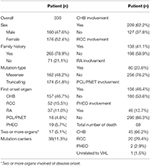
Tabelle 2. Klinische Baseline-Merkmale von Patienten mit VHL-Krankheit.
Univariate und multivariate Cox-Regressionsanalysen für das Gesamtüberleben
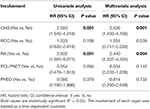
Tabelle 3. Univariate und multivariate Cox-Regressionsanalysen für das Gesamtüberleben.
Auswirkungen einer unterschiedlichen Organbeteiligung auf das CHB-spezifische Überleben
, da CHB fast zwei Drittel des Todes ausmachte (66.2%, 45 von 68) und CHB war ein signifikanter Risikofaktor für das Gesamtüberleben Sowohl in univariaten als auch in multivariaten Cox-Regressionsanalysen untersuchten wir weiter die Wirkung der Beteiligung anderer Organe auf das CHB-spezifische Überleben. Die Beteiligung jedes Organs wurde als zeitabhängige Kovariate behandelt.
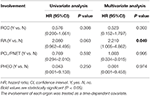
Tabelle 4. Univariate und multivariate zeitabhängige Cox-Regressionsanalysen der Wirkung unterschiedlicher Organbeteiligung auf das CHB-spezifische Überleben.
Die Auswirkung der RCC-Größe auf das Überleben der Patienten
Unsere Datenbank enthielt nur 51 Patienten mit einer vollständigen natürlichen Vorgeschichte von RCC. Wir kategorisierten diese Patienten weiter in 2 Gruppen: die erste Gruppe, RCC-Durchmesser von < 3 cm während ihrer Naturgeschichte (n = 15) und die zweite Gruppe, RCC-Durchmesser von > 3 cm in der Zeit ihrer Naturgeschichte (n = 36). Alle Patienten in der ersten Gruppe überlebten, Drei Patienten in der zweiten Gruppe starben am Ende der Nachsorge; einer starb an einem CHB-bedingten Zustand, und die anderen beiden starben an metastasiertem RCC mit den RCC-Durchmessern von 7 und 8,5 cm zum Zeitpunkt der Diagnose.
In dieser Studie waren CHB und RCC immer noch die Haupttodesursachen bei VHL-Patienten und verursachten zusammen 95,6% aller Todesfälle. Wir analysierten die Wirkung jedes beteiligten Organs auf das Gesamtüberleben und das CHB-spezifische Überleben von Patienten mit VHL-Krankheit. CHB und RA waren unabhängige Risikofaktoren für das Gesamtüberleben. Überraschenderweise war RCC kein signifikanter Risikofaktor für das Gesamtüberleben. RA war der einzige Risikofaktor für das CHB-spezifische Überleben. In den letzten Jahrzehnten hat sich die Todesursache bei Patienten mit VHL-Krankheit stark verschoben. In Studien zur VHL-Krankheit, die in den Anfangsjahren veröffentlicht wurden, trugen CHB und RCC ungefähr zu gleichen Teilen zum Tod der Patienten bei; In einigen Studien war die Zahl der Todesfälle aufgrund von RCC sogar doppelt so hoch wie die aufgrund von CHB (29). In neueren Studien war der Anteil der KHK-bedingten Todesfälle jedoch signifikant höher als der der RCC-bedingten Todesfälle (ergänzende Tabelle 1). Diese Diskrepanz kann teilweise durch die Entwicklung fortschrittlicher Bildgebungstechnologien und Behandlungsstrategien erklärt werden, einschließlich gezielter Therapie, minimalinvasiver Chirurgie und aktiver Überwachung für RCCs, Beitrag zur Früherkennung und wirksamen Behandlung vor der Metastasierung von RCCs. Andererseits müssen die pharmakologische und chirurgische Behandlung von CHB weiter verbessert werden.
Obwohl RCC bei VHL-Erkrankungen normalerweise als weniger bösartig angesehen wird und weniger wahrscheinlich metastasiert und ein höheres krebsspezifisches Überleben aufweist als sporadisches klarzelliges RCC (30), ist RCC immer noch eine wichtige Todesursache bei VHL-Patienten. Die meisten Todesfälle sind auf Tumormetastasen und tumorbedingte Komplikationen zurückzuführen. Überraschenderweise war in dieser Studie der Effekt von RCC auf das Gesamtüberleben nicht signifikant. Eine koreanische Studie mit 24 Patienten mit VHL-Krankheit, die 2009 veröffentlicht wurde, zeigte ähnliche Ergebnisse (31). Frühere Studien haben gezeigt, dass der RCC-Durchmesser von > 3 cm ein Risikofaktor für die Metastasierung und das Gesamtüberleben ist (17, 32). In der VHL-Krankheitspatientendatenbank analysierten wir in dieser Studie, unter den 51 Patienten mit vollständiger natürlicher Vorgeschichte der RCC-Tumorgröße, Nur 3 Patienten starben während der Nachsorge, Dies macht es unmöglich, eine Überlebensanalyse durchzuführen. In: Jilg et al. berichtet, dass der mittlere Tumordurchmesser bei Patienten ohne Metastasen signifikant kleiner war als bei Patienten mit Metastasen (33). In: Duffey et al. berichtet im Jahr 2004, dass Patienten mit RCCs kleiner als 3 cm ein geringes Risiko für Fernmetastasen hatten und 3 cm als Cutoff-Wert für chirurgische Eingriffe empfahlen (34). RCC ist immer noch eine wichtige Todesursache bei Patienten mit VHL-Krankheit. Tumorgröße und Wachstumsrate können für die Prognose von größerer Bedeutung sein. Größere Studien mit längeren Nachbeobachtungszeiten sind erforderlich, um die Beziehung zwischen Überleben und RCCs zu untersuchen.
Obwohl CHB als gutartiger Tumor angesehen wird, kann es zu schweren neurologischen Defekten wie Hydrozephalus, Herniation und Hirnstammkompression sowie in seltenen Fällen zu intrakraniellen Blutungen kommen (14, 29). Perioperative / postoperative Komplikationen können auch bei CHB-Patienten zum Tod führen. Die Häufigkeit der CHB-Beteiligung in der Studienprobe kann die Überlebensanalyse beeinflussen. 62,2% (209 von 336) Patienten hatten in unserer Studie KHK, was unter der durchschnittlichen Anzahl von Patienten mit KHK in den in ergänzender Tabelle 1 aufgeführten Studien liegt. CHB entfielen 66.2% aller Todesfallpatienten in dieser Kohorte, was höher ist als 6 der 7 in der ergänzenden Tabelle 1 aufgeführten Studien. Dies kann durch Geburt verursacht werdenJahr der Patienten. In einer dänischen Studie, die 2016 veröffentlicht wurde, steigt die CHB-bedingte Sterblichkeitsrate von 42% (Geburtsjahr 1901-1955, 18 von 50) auf 83% (Geburtsjahr 1956-2010, 5 von 6), wenn das Geburtsjahr zunimmt, mit einer Gesamt-CHB-bedingten Sterblichkeitsrate von 51%. Der Autor führte diese Diskrepanz auf Fortschritte bei der Diagnose und Behandlung von RCC zurück (15). In unserer Studie wurden jedoch 83,3% (280 von 336) Patienten nach 1955 geboren, nur 16,7% (56 von 336)Patienten wurden vor 1955 geboren. Die CHB-bedingte Sterblichkeitsrate betrug 60,1% für Patienten, die vor 1955 geboren wurden, stieg jedoch auf 70,1 und 75% für Patienten, die zwischen 1955 und 1975 und nach 1975 geboren wurden. RA verursacht Blindheit; In unserer Studie war RA jedoch ein signifikanter Risikofaktor für das Gesamtüberleben und das CHB-spezifische Überleben. Die Netzhaut stammt embryologisch aus dem Gehirn, und sowohl CHB als auch RA haben die gleichen histopathologischen Strukturen (7). Die Onkogenese des Hämangioblastoms hängt mit der Dysregulation des HIF-Signalwegs zusammen, die zur Erhöhung des vaskulären endothelialen Wachstumsfaktors und des Erythropoetins führt (35, 36). In: Franke et al. berichtet, dass Deletionsmutationen im VHL-Gen ein höheres Risiko für CHB und RA hatten (37). Daher nehmen wir an, dass RA eine größere Belastung durch CHB widerspiegeln kann, was zu einem ungünstigen Überleben von Patienten mit VHL-Krankheit führt.
Das Hämangioblastom ist ein signifikanter Risikofaktor für das Überleben. Eine weitere mögliche Erklärung dafür ist das früher einsetzende Alter dieses Tumors. Unsere frühere Forschung ergab, dass das früh einsetzende Alter ein Risikofaktor für das Gesamtüberleben und das VHL-krankheitsspezifische Überleben war (18). Das Durchschnittsalter zu Beginn von CHB und RA betrug 28 Jahre.5 bzw. 30 Jahre, wobei die frühesten unter allen VHL-bedingten Läsionen. Wenn CHB und RA als ZNS-Gruppe kombiniert wurden, hatte diese Gruppe auch ein früheres Anfangsalter als diese Gruppe. Wir führten einen Kruskal-Wallis-Test durch, um das beginnende Alter der Läsionen in 5 Organen zu vergleichen, und P < 0,001 deutet auf einen signifikanten Unterschied hin. Wir machten weiter paarweise Vergleiche und der P-Wert wurde nach Bonferroni-Anpassung bewertet, und es wurden signifikante Unterschiede beim Altersbeginn zwischen RA-PCL / PNET, RA-RCC, CHB-PCL / PNET und CHB-RCC gefunden (ergänzende Tabelle 2). Obwohl das Hämangioblastom als gutartiger Tumor angesehen wird, kann sein früher Beginn das Überleben der Patienten mit VHL-Krankheit verschlechtern.
Mehrere frühere Studien, an denen nur symptomatische Patienten teilnahmen, haben möglicherweise verzerrte Ergebnisse, die die Rate der beteiligten Organe und die Wirkung der beteiligten Organe auf das Überleben des Patienten überschätzen (31, 38). Wir verwendeten zuerst die zeitabhängige Cox-Regressionsanalyse, um die Wirkung jedes beteiligten Organs auf das Überleben von Patienten mit VHL-Krankheit zu bewerten. In einer früheren Studie wurde das Gesamtüberleben von der Krankheitsdiagnose bis zum Tod berechnet; asymptomatische Mutationsträger können jedoch später Tumore entwickeln und entsprechen nicht den Anforderungen des proportionalen Hazard-Modells (17).
Diese große retrospektive Studie konzentrierte sich aus klinischer Sicht auf die unterschiedliche Organbeteiligung am Überleben von Patienten mit VHL-Krankheit. Die Lebenserwartung von Patienten mit VHL-Krankheit betrug 66 Jahre. Das Vorhandensein von CHB und RA waren die unabhängigen Risikofaktoren für das Gesamtüberleben. RCC war kein signifikanter Risikofaktor für das Gesamtüberleben. RA war ein Risikofaktor für das CHB-spezifische Überleben. Patienten mit RA sollten aufgrund des höheren Todesrisikos durch KHK sorgfältig aktiv überwacht werden. Zukünftige größere Studien mit längeren Nachbeobachtungszeiten sind erforderlich, um die Wirkung von RCC auf das Überleben von VHL-Patienten zu untersuchen. Diese Ergebnisse können helfen, zukünftige genetische Beratung und klinische Entscheidungsfindung zu leiten.
Erklärung zur Datenverfügbarkeit
Die für diese Studie generierten Datensätze stehen dem entsprechenden Autor auf Anfrage zur Verfügung
Ethikerklärung
Die Studien mit menschlichen Teilnehmern wurden vom Institutional Review Board des Peking University First Hospital (Peking, China) überprüft und genehmigt. Die schriftliche Einverständniserklärung zur Teilnahme an dieser Studie wurde vom gesetzlichen Vormund / den nächsten Angehörigen der Teilnehmer abgegeben.
Autorenbeiträge
KG und LC waren für das Konzept und Design der Studie verantwortlich. BZ und JW befassten sich mit den klinischen Daten. SL und XP führten die statistische Arbeit durch. BH und JZho gepfropft das Manuskript. KM und JZha liefert die Zahlen und Tabellen. Alle Autoren haben das Manuskript überarbeitet.
Förderung
Diese Arbeit wurde durch die Grundlagenforschungsfonds der Zentraluniversitäten gefördert (Zuschuss BMU2018JI002).
Interessenkonflikt
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagung
Die Autoren danken Li Xue-ying von der Statistischen Abteilung des Ersten Krankenhauses der Universität Peking für ihre Unterstützung bei den statistischen Analysen und Bu Ding-Fang vom Medical Experiment Center des Ersten Krankenhauses der Universität Peking für seine technische Unterstützung.
Ergänzungsmaterial
Abkürzungen
CHB, Zentralnervensystem Hämangioblastom; RA, retinales Hämangioblastom; RCC, Nierenzellkarzinom; PHEO, Phäochromozytom; PCL / PNET, pankreatische zystische Läsion und pankreatische neuroendokrine Tumoren.
7. Webster AR, Maher ER, Moore BEI. Klinische Merkmale der okulären Angiomatose bei der von-Hippel-Lindau-Krankheit und Korrelation mit der Keimbahnmutation. Arch Ophthalmol. (1999) 117:371–8. doi: 10.1001/archopht.117.3.371
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
10. Er ist der Name folgender Personen: Wanebo JE, Lonser RR, Glenn GM, Oldfield EH. Die Naturgeschichte von Hämangioblastomen des Zentralnervensystems bei Patienten mit von-Hippel-Lindau-Krankheit. J Neurochirurg. (2003) 98:82–94. Ursprungsbezeichnung: 10.3171/jns.2003.98.1.0082
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
13. Webster AR, Richards FM, MacRonald FE, Moore BEI, Maher ER. Eine Analyse der phänotypischen Variation des familiären Krebssyndroms von Hippel-Lindau-Krankheit: Evidenz für Modifikatoreffekte. Bin J Hum Genet. (1998) 63:1025–35. doi: 10.1086/302037
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
19. El-Galaly TC, Cheah CY, Hutchings M, Mikhaeel NG, Savage KJ, Sehn LH, et al. Uterine, aber nicht ovarielle, weibliche Fortpflanzungsorganbeteiligung bei Präsentation durch diffuses großes B-Zell-Lymphom ist mit schlechten Ergebnissen und einer hohen Häufigkeit einer sekundären ZNS-Beteiligung verbunden. Br J Haematol. (2016) 175:876–83. Ursprungsbezeichnung: 10.1111/bjh.14325
Querverweis Volltext / Google Scholar
20. Yuda S, Hayashi T, Yasui K, Muranaka A, Ohnishi H, Hashimoto A, et al. Perikarderguss und Beteiligung mehrerer Organe sind unabhängige Prädiktoren für die Mortalität bei Patienten mit systemischer Leichtkettenamyloidose. In: Intern Med. (2015) 54:1833–40. doi: 10.2169/innere Medizin.54.3500
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
27. Wu P., Zhang N., Wang X., Ning X., Li T., Bu D., et al. Die Familienanamnese der von-Hippel-Lindau-Krankheit war bei chinesischen Patienten ungewöhnlich: Dies deutet auf die höhere Häufigkeit von De-novo-Mutationen im VHL-Gen bei diesen Patienten hin. J Hum Genet. (2012) 57:238–43. Ursprungsbezeichnung: 10.1038/jhg.2012.10
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
30. Neumann HP, Bender BU, Berger DP, Laubenberger J, Schultze-Seemann W, Wetterauer U, et al. Prävalenz, Morphologie und Biologie des Nierenzellkarzinoms bei von-Hippel-Lindau-Krankheit im Vergleich zum sporadischen Nierenzellkarzinom. J Urol. (1998) 160:1248–54. ust-idnr.: 10.1016/S0022-5347(01)62509-6
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
31. Kim WT, Schinken WS, Ju HJ, Lee JS, Lee JS, Choi YD. Klinische Merkmale des Nierenzellkarzinoms bei koreanischen Patienten mit von-Hippel-Lindau-Krankheit im Vergleich zu sporadischem bilateralem oder multifokalem Nierenzellkarzinom. In: J Korean Med Sci. (2009) 24:1145–9. geburtsdatum: 10.3346/jkms.2009.24.6.1145
Querverweis Volltext / Google Scholar
35. Wizigmann-Voos S, Breier G, Risau W, Platte KH. Hochregulation des vaskulären endothelialen Wachstumsfaktors und seiner Rezeptoren bei von-Hippel-Lindau-assoziierten und sporadischen Hämangioblastomen. Cancer Res. (1995) 55:1358-64.
PubMed Zusammenfassung / Google Scholar
36. Krieg M, Marti HH, Platte KH. Koexpression von Erythropoetin und vaskulärem endothelialem Wachstumsfaktor bei Tumoren des Nervensystems im Zusammenhang mit Funktionsverlust des von Hippel-Lindau-Tumorsuppressorgens. Blut. (1998) 92:3388–93.
PubMed Zusammenfassung / Google Scholar
37. Franke G, Bausch B, Hoffmann MM, Cybulla M, Wilhelm C, Kohlhase J, et al. Die Alu-Alu-Rekombination liegt der überwiegenden Mehrheit der großen VHL-Keimbahndeletionen zugrunde: molekulare Charakterisierung und Genotyp-Phänotyp-Korrelationen bei VHL-Patienten. Hum Mutat. (2009) 30:776–86. doi: 10.1002/humu.20948
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar