- colectarea probelor, construcția bibliotecii și secvențierea
- controlul calității
- estimarea dimensiunii genomului
- asamblarea și evaluarea genomului
- adnotare repetitivă a elementelor
- adnotarea genelor care codifică proteinele
- adnotarea funcției genetice
- construcția familiei de Gene și reconstrucția filogenezei
colectarea probelor, construcția bibliotecii și secvențierea
ADN Genomic a fost obținut dintr-un specimen masculin de C. crocuta (NCBI taxonomy ID: 9678; Fig. 1) depozitat în grădina zoologică înghețată de la Institutul Zoologic din San Diego pentru cercetări de conservare, SUA (ID-ul grădinii zoologice înghețate: KB4526).
ADN-ul genomic a fost extras folosind fenol-cloroform urmat de purificare folosind precipitarea etanolului13. ADN-ul extras a fost rulat și vizualizat pe un 1.5% gel de agaroză rulează în tampon 1x TBE pentru a verifica prezența ADN-ului cu greutate moleculară mare. Concentrația și puritatea ADN-ului au fost cuantificate pe un spectrofotometru NanoDrop 2000 și Fluorometru Qubit 2.0 (Thermo Fisher Scientific, SUA) înainte de expediere la BGI-Shenzhen, China. S–a obținut un total de 372 de hectolitri de ADN genomic, cu o concentrație de 0,418 de centimetrii/centimetrii folosind Nanodrop 2000 și 0,245-0,399 de centimetrii/centimetrii pe baza a patru citiri replicate folosind Fluorometrul Qubit 2,0. Raportul de puritate 260/280 a fost de 1,95. Apoi am codificat proba folosind gena citocromului b (Cytb). Apoi, conform strategiei gradient library, am construit 13 biblioteci de dimensiuni inserate, cu următoarele lungimi de dimensiuni inserate: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Am folosit HiSeq. 2000 sequencer (Illumina, SUA) pentru a sequence Paired-End (PE) Citește pentru fiecare bibliotecă pe 14 benzi. Un total de aproximativ 299 Gb date brute au fost generate din 13 biblioteci, obținând o adâncime de secvențiere (acoperire) de 149,25 (Tabelul 1).
controlul calității
pentru a minimiza erorile de asamblare greșită, am filtrat citirile brute înainte de asamblarea genomului de novo conform următoarelor două criterii. În primul rând, au fost eliminate citirile cu mai mult de 10 bp aliniate la secvența adaptorului (permițând <= nepotrivire de 3 bp). În al doilea rând, citirile cu 40% din bazele care au o valoare de calitate mai mică sau egală cu 10 au fost aruncate. În cele din urmă, am obținut 190,4 g date cu o acoperire de 95,2 (Tabelul 2).
estimarea dimensiunii genomului
trei biblioteci cu inserție scurtă (două de 170 bp și una de 500 bp) au fost utilizate pentru a estima dimensiunea genomului și heterozigozitatea la nivel de genom prin analiza k-mer. Un total de aproximativ 385 m pe citiri au fost trimise la jellyfish14 pentru a calcula frecvența k-mer. Apoi distribuția k-mer a fost ilustrată de Genomescope7 cu parametrii”k = 17; Lungime = 100; acoperire maximă = 1000″. Am obținut o dimensiune estimată a genomului 2,003,681,234 bp și heterozigozitate de 0,325% (Fig. 2).

17-estimarea mer a dimensiunii genomului. Axa x este adâncimea (X), axa y este proporția care reprezintă frecvența la acea adâncime împărțită la frecvența totală a tuturor adâncimilor de acoperire. Fără luarea în considerare a ratei de eroare a secvenței, a ratei de heterozigozitate și a ratei de repetare a genomului, distribuția de 17 mer ar trebui să aproximeze o distribuție Poisson.
asamblarea și evaluarea genomului
SOAPdenovo (V1.06) 15 a fost folosit pentru asamblarea genomului de novo, în urma filtrării datelor de dimensiune scurtă a inserției și eliminarea vârfului mic al datelor de dimensiune mare a inserției. Algoritmul de asamblare SOAPdenovo a inclus trei pași principali. (1) construcție Contig: datele bibliotecii de dimensiuni scurte au fost împărțite în K-mers și construite folosind un grafic de Bruijn, care a fost simplificat prin eliminarea vârfurilor, fuzionarea bulelor, eliminarea acoperirii reduse a conexiunii și eliminarea repetărilor mici. Am obținut secvența contig prin conectarea căii k-mer, rezultând un contig N50 2,104 bp și lungimea totală 2,295,545,898 bp. (2) construcția schelei: am obținut 80% din toate citirile aliniate cu capăt pereche prin realinierea tuturor citirilor utilizabile pe contigs. Apoi am calculat cantitatea de relații pereche partajate între fiecare pereche de contiguri, am ponderat rata capetelor pereche consistente și conflictuale și apoi am construit schelele pas cu pas. Ca rezultat, am obținut schele cu un N50 7,168,038 bp și lungimea totală 2,355,303,269 bp de la capete pereche scurte de dimensiuni inserate, până la capete pereche îndepărtate. (3) închiderea Gap-ului: pentru a umple golurile din interiorul schelelor construite, am folosit informațiile pereche-end pentru a prelua perechile citite pentru a face din nou un ansamblu local pentru aceste citiri colectate. Pe scurt, am închis 87,7% din golurile intra-schele sau 85,8% din lungimea gap-ului sumei. Dimensiunea contig N50 a crescut de la 2.104 bp la 21.301 bp (Tabelul 3). Dimensiunea ansamblului schelei a fost de 2.355.303.269 bp, care este aproape de dimensiunea genomului bazată pe asamblare de 2.374.716.107 BP raportată pentru hyaena dungată, Hyaena hyaena11 (aderarea NCBI: ASM300989v1). De asemenea, am recuperat și adnotat genomul mitocondrial al hienei reperate folosind programul Mitoz16, care are o lungime de 16.858 bp, similar cu primii genomi mitocondriali secvențiați pentru această specie12.
evaluarea proiectului genomului a fost efectuată analizând completitudinea ortologilor cu o singură copie folosind BUSCO (versiunea 3.1.0)17, căutând în baza de date Mammaliaodb9 care conține 4.104 grupuri ortologice cu o singură copie. Un total de 95,5% dintre ortologi au fost identificați ca fiind complete, 2,5% ca fragmentate și 2,0% ca lipsă, indicând o calitate generală ridicată a ansamblului genomului hienei reperate. Având în vedere că 99,95% din schelele scurte (<1k) adăposteau doar 1.2% din lungimea totală a genomului, am exclus aceste schele pentru analiza în aval, inclusiv elemente repetitive și adnotare caracteristică genică.
adnotare repetitivă a elementelor
atât repetițiile în tandem, cât și elementele transpozabile (te) au fost căutate și identificate în genomul C. crocuta. Repetările Tandem au fost identificate folosind Căutătorul de repetări Tandem (TRF, v4.07)18 și elementele transpozabile (TEs) au fost identificate printr-o combinație de abordări bazate pe omologie și de novo. Pentru predicția bazată pe omologie, am folosit versiunea RepeatMasker 4.0.619 cu setările „- nolow-no_is-norna-engine ncbi ” și RepeatProteinMask (un program din pachetul RepeatMasker) cu setările „-engine ncbi-nolowsimple-pvalue 0.0001” pentru a căuta TEs la nivelul nucleotidelor și aminoacizilor pe baza repetărilor cunoscute (Fig. 3). RepeatMasker a fost aplicat pentru identificarea la nivel de ADN folosind o bibliotecă personalizată care a combinat repbase21.10 dataset20. La nivelul proteinei, RepeatProteinMask a fost utilizat pentru a efectua RMBlast împotriva bazei de date de proteine TE. Pentru predicția ab initio, RepeatModeler (v1.0.8)21 și LTR_FINDING (v1.06) 22 au fost aplicate pentru a construi biblioteca de novo repeat. Secvențele de contaminare și copiere multiplă din bibliotecă au fost eliminate, iar secvențele rămase au fost clasificate în funcție de rezultatul exploziei în urma alinierii la baza de date SwissProt. Pe baza acestei biblioteci, am folosit RepeatMasker pentru a masca tes omoloage și le-a clasificat (Fig. 4). În general, un total de 826 Mb de elemente repetitive au fost identificate în hiena reperată, cuprinzând 35,29% din întregul genom (Tabelul 4).

distribuția ratei de divergență a fiecărui tip de element transpozabil (TE) în ansamblul genomului Crocuta crocuta pe baza predicției bazate pe omologie. Rata de divergență a fost calculată între TEs identificat în genom folosind o metodă bazată pe omologie și secvența de consens din Baza de date Repbase 20.

distribuția ratei de divergență a fiecărui tip de te în ansamblul genomului Crocuta crocuta bazat pe predicția ab initio. Rata de divergență a fost calculată între TEs identificat în genom prin predicția ab initio și secvența de consens din biblioteca te prezisă (vezi metode).
adnotarea genelor care codifică proteinele
am folosit predicția ab initio și abordările bazate pe omolog pentru a adnota genele care codifică proteinele, precum și site-urile de îmbinare și izoformele alternative de îmbinare. Predicția Ab initio a fost efectuată pe genomul mascat repetat folosind modele de gene de la câine uman, domestic și pisică domestică folosind AUGUSTUS (versiunea 2.5.5)23, GENSCAN24, GlimmerHMM (versiunea 3.0.4)25 și SNAP (versiunea 2006-07-28)26, respectiv. Un total de 22.789 de gene au fost identificate prin această metodă. Proteinele omoloage ale, Homo sapiens, Felis catus și Canis familiaris (din eliberarea Ensembl 96)au fost mapate la genomul hienei reperate folosind tblastn (Blastall 2.2.26) 27 cu parametrii „-e 1e-5”. Secvențele aliniate, precum și proteinele lor de interogare au fost apoi trimise la GeneWise (versiunea 2.4.1)28 pentru căutarea unei alinieri precise îmbinate. Setul final de gene (22.747) a fost colectat prin fuzionarea rezultatelor ab initio și homolog folosind o conductă personalizată (Tabelul 5).
adnotarea funcției genetice
funcțiile genetice au fost atribuite în funcție de cea mai bună potrivire obținută prin alinierea secvențelor de codificare a genelor traduse folosind BLASTP cu parametrii „-e 1e-5” la bazele de date SwissProt și TrEMBL (Uniprot release 2017-09). Motivele și domeniile genelor au fost determinate de InterProScan (v5)29 împotriva bazelor de date proteice, inclusiv ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 și PROSITE35. ID-urile de Ontologie genetică pentru fiecare genă au fost obținute din intrările SwissProt și TrEMBL corespunzătoare. Toate genele au fost aliniate împotriva proteinelor KEGG, iar calea în care gena ar putea fi implicată a fost derivată din genele potrivite din Baza de date KEGG36. În rezumat, 22.166 (97,45%) din genele prezise de codificare a proteinelor au fost adnotate cu succes de cel puțin una dintre cele șase baze de date (Tabelul 6).
construcția familiei de Gene și reconstrucția filogenezei
pentru a obține o perspectivă asupra istoriei filogenetice și a evoluției familiilor de gene ale Crocuta crocuta, am grupat secvențe de gene din șapte specii (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) și Homo sapiens ca outgroup (Ensembl release-96, Panthera leo din date nepublicate) în gene familii care utilizează ortomcl (v2.0.9)37. Genele care codifică proteinele pentru cele opt specii au fost recuperate prin selectarea celei mai lungi izoforme de transcriere pentru fiecare genă pentru atribuirea perechilor în aval (construirea grafurilor). Am efectuat o căutare BLASTP all-contra-all pe secvențele de proteine ale tuturor speciilor de referință, cu o limită de valoare E de 1e-5. Construcția familiei de Gene a folosit algoritmul MCL38 cu parametrul de inflație de ‘1,5’. Un total de 16.271 de familii de gene de C. crocuta, H. sapiens, F. catus, A. melanoleuca au fost grupate. Au existat 11.671 de familii de gene împărtășite de aceste patru specii, în timp ce 292 de familii de gene care conțineau 1.446 de gene erau specifice C. Crocuta (Fig. 5). În mod evident, familiile de gene C. crocuta și F. catus împărtășite au fost mai mici decât C. crocuta și H. sapiens împărtășite, ceea ce ar putea rezulta din faptul că H. sapiens avea un genom și o adnotare mai complete.
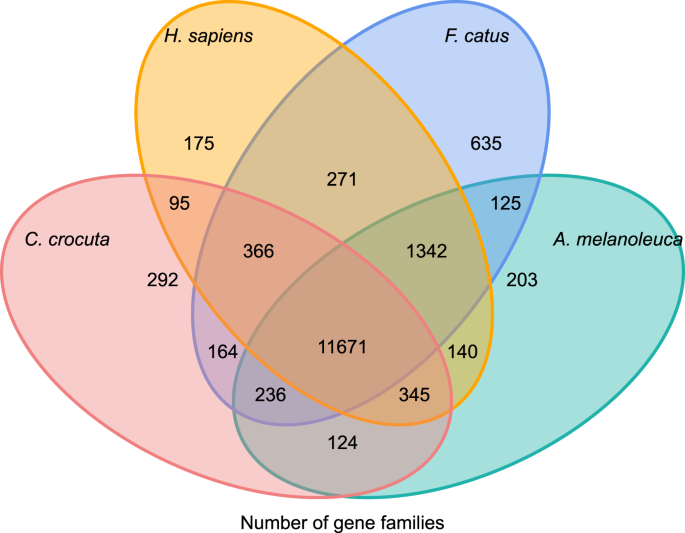
Diagrama Venn care arată Compararea genelor comune și unice de codificare a proteinelor între hiena reperată, om, pisică domestică și câine domestic pe baza analizei ortologice.
am identificat 6.601 gene ortologice cu o singură copie pentru a reconstrui arborele filogenetic al celor opt specii. Aliniamentele multiple ale secvențelor de aminoacizi pentru fiecare genă au fost generate folosind mușchi (versiunea 3.8.31)39 și tăiate folosind Gblocks (0.91 b)40, obținând regiuni bine aliniate cu parametrii „-t = p-b3 = 8-b4 = 10-b5 = n-E = – st”. Am efectuat analiza filogenetică utilizând metoda probabilității maxime, implementată în PhyML (v3.0)41, utilizând modelul JTT + G + I pentru substituția aminoacizilor (Fig. 6). Rădăcina arborelui a fost determinată prin minimizarea înălțimii întregului copac prin Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). În cele din urmă, am estimat timpul de divergență între cele opt linii folosind MCMCTree din pachetul software PAML versiunea 4.442. Pentru calibrarea ratei de substituție au fost utilizate două antecedente bazate pe înregistrarea fosilelor, inclusiv Boreoutheria (91-102 MYA) și Carnivora (52-57 Mya)43. În concordanță cu studiile anterioare, grupurile de hienă reperată cu cele patru specii incluse din Felidae într-o cladă care definește subordinea Feliformia, care s-a abătut de la Caniformia (reprezentată de câinele domestic și panda gigant) 53,9 Mya44.

arborele filogenetic al lui C. crocuta și alte șapte specii construite prin metoda probabilității maxime pe baza a 6.601 ortologi cu o singură copie. Timpul de divergență a fost estimat folosind cele două priorități de calibrare derivate din Baza de date Time Tree (http://www.timetree.org), care sunt marcate de un romb roșu. Toate timpii estimați de divergență sunt afișați cu intervale de încredere de 95% între paranteze.