Prøveinnsamling, bibliotek konstruksjon og sekvensering
Genomisk DNA ble oppnådd fra en mannlig prøve Av C. crocuta (NCBI taksonomi ID: 9678; Fig. 1) lagret I frozen Zoo® Ved San Diego Zoo Institute For Conservation Research, USA (Frozen Zoo ID: KB4526).
det genomiske DNA ble ekstrahert ved bruk av fenol-kloroform etterfulgt av rensing ved bruk av etanol utfelling 13. Det ekstraherte DNA ble kjørt og visualisert på en 1.5% agarose gel kjøre i 1x TBE buffer for å sjekke for TILSTEDEVÆRELSE AV HØY molekylvekt DNA. DNA konsentrasjon og renhet ble kvantifisert På Et NanoDrop 2000 spektrofotometer og Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA) før frakt til Bgi-Shenzhen, Kina. Vi oppnådde totalt 372 µ av genomisk DNA, med en konsentrasjon på 0.418@g/µ ved Bruk Av Nanodrop 2000 og 0.245–0.399@g/hryvnas basert på fire replikatavlesninger ved Bruk Av Qubit 2.0-Fluorometeret. 260/280 forholdet mellom renhet var 1,95. Vi deretter barcoded prøven ved hjelp av cytokrom b (Cytb) gen. Deretter konstruerte vi i henhold til gradientbibliotekstrategien 13 sett inn-størrelse biblioteker, med følgende innsatsstørrelseslengder: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Vi brukte HiSeq. 2000 sequencer (Illumina, USA) til sequence Paired-End (PE) leser for hvert bibliotek over 14 baner. 299 Gb rådata ble generert fra 13 biblioteker, og oppnådde en sekvenseringsdybde (dekning) på 149,25 (Tabell 1).
Kvalitetskontroll
for å minimere feilmonteringsfeil filtrerte vi raw-leser før de novo genommontering i henhold til følgende to kriterier. Først leser med mer enn 10 bp justert til adaptersekvensen (slik at < = 3 bp mismatch) ble fjernet. Sekund, leser med 40% av baser som har en kvalitet verdi mindre enn eller lik 10 ble forkastet. Til slutt fikk vi 190,4 g data med en dekning på 95,2 (Tabell 2).
Estimering av genomstørrelse
Tre kortinnsatsbiblioteker (to av 170 bp og en av 500 bp) ble brukt til å estimere genomstørrelsen og genom-bred heterozygositet ved k-mer analyse. Totalt ca 385 M PE leser ble sendt til jellyfish14 å beregne k – mer frekvens. Deretter ble k-mer-distribusjonen illustrert Av Genomescope7 med parametere «k = 17; lengde = 100; maks dekning = 1000». Vi oppnådde en estimert genomstørrelse 2,003,681,234 bp, og heterozygositet på 0,325% (Fig. 2).

17-mer estimat av genom størrelse. X-aksen er dybde (X), y-aksen er andelen som representerer frekvensen på den dybden dividert med den totale frekvensen av alle dekningsdybder. Uten hensyn til sekvensfeilfrekvensen, heterozygositetshastigheten og repetisjonshastigheten av genomet, bør 17-mer-fordelingen tilnærmet en Poisson-fordeling.
Genom montering og vurdering
SOAPdenovo (V1.06) 15 ble ansatt for å montere genomet de novo, etter filtrering av de korte innsatsstørrelsesdataene og fjerne den lille toppen av de store innsatsstørrelsesdataene. SOAPdenovo-monteringsalgoritmen inneholdt tre hovedtrinn. (1) Contig construction: bibliotekets kortinnsatsstørrelsesdata ble delt inn i k-mers og konstruert ved hjelp av en De Bruijn-graf, som ble forenklet ved å fjerne tips, slå sammen bobler, fjerne lav dekning av forbindelsen og fjerne små gjentakelser. Vi oppnådde contig-sekvensen ved å koble k-mer-banen, noe som resulterte i en contig N50 2,104 bp, og total lengde 2,295,545,898 bp. (2) Stillaskonstruksjon: vi oppnådde 80% av alle justerte parrede leser ved å justere alle brukbare les på contigs. Deretter beregnet vi mengden delte sammenkoblede relasjoner mellom hvert par kontigs, veide frekvensen av konsistente og motstridende sammenkoblede ender, og konstruerte deretter stillasene trinnvis. Som et resultat fikk vi stillas med En N50 7,168,038 bp, og total lengde 2,355,303,269 bp fra korte innsatsstørrelser parede ender til lange fjerne parede ender. (3) Gap closing: for å fylle hullene i de konstruerte stillasene brukte vi den sammenkoblede informasjonen til å hente leseparene for å gjøre en lokal montering igjen for disse innsamlede lesene. I sammendraget lukkede vi 87.7% av intra-stillasgapene, eller 85.8% av summen gap lengde. Contig n50-størrelsen økte fra 2.104 bp til 21.301 bp (Tabell 3). Stillasmonteringsstørrelsen var 2,355,303,269 bp, som ligger nær den monteringsbaserte genomstørrelsen på 2,374,716,107 bp rapportert for den stripete hyaena, Hyaena hyaena11 (NCBI tiltredelse: ASM300989v1). Vi hentet også og annoterte mitokondriellgenomet av den spotted hyena ved Hjelp Av MitoZ-programmet16, som har en lengde på 16.858 bp, ligner de første mitokondrielle genomene sekvensert for denne arten12.
Vurdering av utkastet genomet ble utført ved å se på fullstendigheten av single-copy orthologs bruker BUSCO (versjon 3.1.0)17, søker Mot Mammaliaodb9 database som inneholder 4104 single-copy ortholog grupper. Totalt 95,5% av ortologene ble identifisert som komplette, 2,5% som fragmenterte og 2,0% som manglende, noe som indikerer en samlet høy kvalitet på den flekkete hyengenomenheten. Gitt at 99,95% av de korte stillasene (< 1k)bare hadde 1.2% av den totale genomlengden, ekskluderte vi disse stillasene for nedstrøms analyse, inkludert repeterende element og genfunksjonsannotasjon.
Repeterende element annotasjon
både tandem gjentar og transposable elementer (TE) ble søkt etter Og identifisert over C. crocuta genomet. Tandem repetisjoner ble identifisert Ved Hjelp Av Tandem Gjentar Finder (TRF, v4.07)18 og transposable elementer (TEs) ble identifisert ved en kombinasjon av homologi-baserte og de novo tilnærminger. For den homologibaserte prediksjonen brukte Vi RepeatMasker versjon 4.0.619 med innstillingene «- nolow-no_is-norna-engine ncbi «Og RepeatProteinMask (et program Innen RepeatMasker pakke) med innstillingene»- engine ncbi-noLowSimple-pvalue 0.0001 » for å søke TEs på nukleotid og aminosyre nivå basert på kjente repetisjoner (Fig. 3). RepeatMasker ble søkt OM DNA-nivåidentifikasjon ved hjelp av et tilpasset bibliotek som kombinerte Repbase21. 10 dataset20. På proteinnivået Ble RepeatProteinMask brukt til å utføre RMBlast mot te-proteindatabasen. For ab initio prediksjon, RepeatModeler (v1. 0.8)21 og LTR_FINDING (v1.06) 22 ble brukt til å konstruere de novo repeat biblioteket. Forurensning og multi-kopi sekvenser i biblioteket ble fjernet og de resterende sekvenser ble klassifisert i henhold TIL BLAST resultat etter justering Til SwissProt database. Basert på dette biblioteket brukte Vi RepeatMasker til å maskere de homologe TEs og klassifisere dem (Fig. 4). Totalt ble det identifisert 826 Mb repeterende elementer i flekket hyene, som utgjorde 35,29% av hele genomet (Tabell 4).

Fordeling av divergens rate av hver type transposable element (TE) I Crocuta crocuta genomet montering basert på homologi – basert prediksjon. Divergensraten ble beregnet mellom de identifiserte TEs i genomet ved hjelp av en homologibasert metode og konsensussekvensen i Repbase-databasen20.

Fordeling av divergens rate av HVER TYPE TE I Crocuta crocuta genomet montering basert på ab initio prediksjon. Divergensraten ble beregnet mellom de identifiserte TEs i genomet ved ab initio-prediksjon og konsensussekvensen i det forutsagte TE-biblioteket (se Metoder).
Proteinkodende genannotasjon
Vi brukte ab initio-prediksjon og homologbaserte tilnærminger til å annotere proteinkodende gener samt spleisingssteder og alternative spleisingsisoformer. Ab initio prediksjon ble utført på repeat-maskert genom ved hjelp av genmodeller fra menneske, husdyr, og huskatt VED HJELP AV AUGUSTUS (versjon 2.5.5)23, GENSCAN24, GlimmerHMM (versjon 3.0.4)25, OG SNAP (versjon 2006-07-28)26, henholdsvis. Totalt 22 789 gener ble identifisert ved denne metoden. Homologe proteiner Av, Homo sapiens, Felis catus Og Canis familiaris (Fra Ensembl 96-utgivelsen) ble kartlagt til det spotted hyena genomet ved hjelp av tblastn (Blastall 2.2.26) 27 med parametere «- e 1e-5». De justerte sekvensene samt deres spørringsproteiner ble deretter sendt Til GeneWise (versjon 2.4.1) 28 for å søke en nøyaktig spleiset justering. Det endelige gensettet (22 747) ble samlet inn ved å slå sammen ab initio og homolog-baserte resultater ved hjelp av en tilpasset rørledning (Tabell 5).
genfunksjonsannotasjon
Genfunksjoner ble tildelt i henhold til den beste matchen oppnådd ved å tilpasse oversatte genkodingssekvenser ved HJELP AV BLASTP med parametere «- e 1e-5 » Til SwissProt og TrEMBL-databasene (Uniprot release 2017-09). Motiver og domener av gener ble bestemt Av InterProScan (v5) 29 mot protein databaser Inkludert ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 OG PROSITE35. Gene Ontology IDs for hvert gen ble oppnådd fra de tilsvarende SwissProt og TrEMBL-oppføringene. Alle gener ble justert mot kegg-proteiner, og banen der genet kan være involvert, ble avledet fra de matchede gener i kegg-databasen36. Oppsummert ble 22.166 (97.45%) av de forventede proteinkodende generene vellykket annotert av minst en av de seks databasene (Tabell 6).
Genfamiliekonstruksjon og fylogeni rekonstruksjon
For å få innsikt i Den fylogenetiske historien Og utviklingen Av genfamilier Av Crocuta crocuta, grupperte vi gensekvenser av syv arter (Felis catus, Canis familiaris, ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) og Homo sapiens som utgruppe (Ensembl release-96, Panthera leo fra upubliserte data) til Genfamilier ved hjelp av orthomcl (v2.0.9)37. De proteinkodende genene for de åtte artene ble hentet ved å velge den lengste transkriptisoformen for hvert gen for nedstrøms parvis tildeling (grafbygging). Vi utførte EN all-mot-ALLE BLASTP søk på protein sekvenser av alle referansearter, Med En e-verdi cut-off av 1e-5. Gene family construction ansatt MCL algorithm38 med inflasjonsparameteren ‘1.5’. Totalt 16.271 genfamilier Av c. crocuta, h. sapiens, F. catus, a. melanoleuca ble gruppert. Det var 11.671 genfamilier delt av disse fire artene, mens 292 genfamilier som inneholdt 1.446 gener var spesifikke For C. Crocuta (Fig. 5). Merkbart var genfamiliene c. crocuta og f. catus delt mindre enn c. crocuta og H. sapiens delt, noe Som kunne skyldes At h. sapiens hadde et mer komplett genom og merknad.
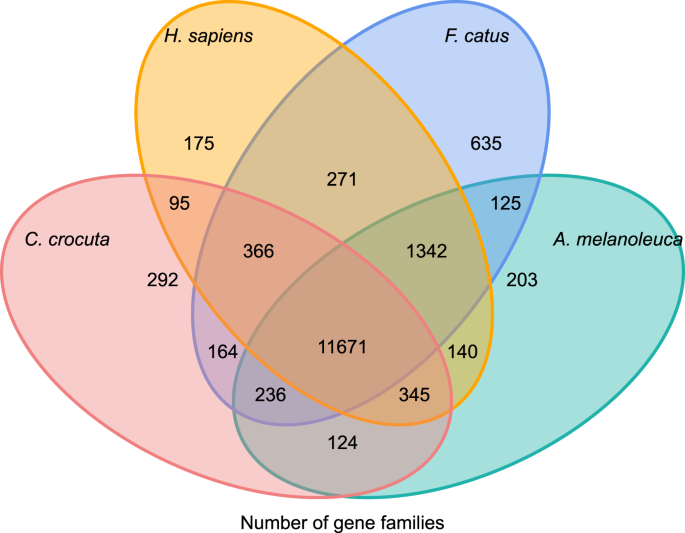
Venn-diagram som viser sammenligning av delte og unike proteinkodende gener blant spotted hyena, human, domestic cat og husdyr basert på ortologianalyse.
Vi identifiserte 6,601 enkeltkopierte orthologe gener for å rekonstruere det fylogenetiske treet av de åtte artene. Flere sekvensjusteringer av aminosyresekvenser for hvert gen ble generert VED HJELP AV MUSKEL (versjon 3.8.31)39, og trimmet ved Hjelp Av Gblocks (0.91 b)40, og oppnådde veljusterte regioner med parametrene «-t = p-b3 = 8-b4 = 10-b5 = n-e = – st». Vi utførte fylogenetisk analyse ved hjelp av maksimalsannsynlighetsmetoden som implementert I PhyML (v3.0)41, ved BRUK AV jtt + G + i-modellen for aminosyresubstitusjon (Fig. 6). Roten av treet ble bestemt ved å minimere høyden på hele treet via Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Til slutt estimerte vi divergenstiden blant de åtte linjene ved Hjelp Av MCMCTree fra PAML versjon 4.4 programvarepakke42. To priorer basert på fossilene ble brukt til å kalibrere substitusjonsraten, inkludert Boreoeutheria (91-102 MYA) og Carnivora (52-57 MYA)43. I samsvar med tidligere studier, spotted hyena grupper med de fire artene inkludert Fra Felidae i en klade definere underordenen Feliformia, som skilte Seg fra Caniformia (representert ved den innenlandske hunden Og kjempepandaen) 53,9 Mya44.

Fylogenetisk tre Av c. crocuta og syv andre arter konstruert av maksimal sannsynlighetsmetode basert på 6,601 enkeltkopierte ortologer. Divergenstiden ble estimert ved hjelp av de to kalibreringspriorene avledet fra Time Tree-databasen (http://www.timetree.org), som er merket med en rød rhombus. Alle estimerte divergenstider er vist med 95% konfidensintervall i parentes.