provsamling, bibliotekskonstruktion och sekvensering
genomiskt DNA erhölls från ett manligt prov av C. crocuta (NCBI taxonomi ID: 9678; Fig. 1) lagrad i den frusna djurparken på San Diego Zoo Institute for Conservation Research, USA (fryst Zoo-ID: KB4526).
det genomiska DNA extraherades med användning av fenol-kloroform följt av rening med användning av etanolutfällning13. Det extraherade DNA kördes och visualiserades på en 1.5% agarosgel körs i 1x TBE-buffert för att kontrollera närvaron av DNA med hög molekylvikt. DNA-koncentration och renhet kvantifierades på en NanoDrop 2000 spektrofotometer och Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA) före leverans till BGI-Shenzhen, Kina. Vi erhöll totalt 372 Baccarat av genomiskt DNA, med en koncentration av 0.418 Baccarat / Baccarat med användning av Nanodrop 2000 och 0.245 – 0.399 Baccarat/Baccarat baserat på fyra replikat avläsningar med användning av Qubit 2.0 Fluorometer. Förhållandet 260/280 av renhet var 1,95. Vi streckkodade sedan provet med cytokrom b (Cytb) gen. Sedan, enligt gradient library-strategin, konstruerade vi 13 bibliotek med insatsstorlek, med följande insatsstorlekslängder: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Vi använde HiSeq. 2000 sequencer (Illumina, USA) för att sekvensera Paired-End (PE) läser för varje bibliotek över 14 banor. Totalt cirka 299 Gb rådata genererades från 13 bibliotek, vilket uppnådde ett sekvenseringsdjup (täckning) på 149,25 (Tabell 1).
kvalitetskontroll
för att minimera felmonteringsfel filtrerade vi raw-läsningar före de novo genommontering enligt följande två kriterier. Först läser med mer än 10 bp i linje med adaptersekvensen (tillåter <= 3 bp mismatch) avlägsnades. För det andra, läser med 40% av baser som har ett kvalitetsvärde mindre än eller lika med 10 kasserades. Slutligen erhöll vi 190.4 G-data med en täckning av 95.2 (Tabell 2).
uppskattning av genomstorlek
tre kortinsatsbibliotek (två av 170 bp och en av 500 bp) användes för att uppskatta genomstorleken och genombrett heterozygositet genom k-Mer-analys. Totalt cirka 385 m PE-läsningar skickades till jellyfish14 för att beräkna k-Mer-frekvensen. Sedan illustrerades k-Mer-distributionen av Genomescope7 med parametrarna ”k = 17; längd = 100; max täckning = 1000”. Vi erhöll en uppskattad genomstorlek 2,003,681,234 bp och heterozygositet på 0,325% (Fig. 2).

17-mer uppskattning av genomstorlek. X-axeln är djup (X), y-axeln är den andel som representerar frekvensen vid det djupet dividerat med den totala frekvensen för alla täckningsdjup. Utan hänsyn till sekvensfelfrekvensen, heterozygositetshastigheten och upprepningshastigheten för genomet, bör 17-mer-fördelningen approximera en Poisson-fördelning.
genommontering och bedömning
SOAPdenovo (V1.06) 15 användes för att montera genomet de novo, efter filtrering av de korta insatsstorleksdata och avlägsnande av den lilla toppen av de stora insatsstorleksdata. SOAPdenovo-monteringsalgoritmen inkluderade tre huvudsteg. (1) Contig konstruktion: kort-infoga storlek biblioteksdata delades upp i k-mers och konstrueras med användning av en de Bruijn graf, som förenklades genom att ta bort tips, sammanslagning bubblor, ta bort den låga täckningen av anslutningen och ta bort små upprepningar. Vi erhöll contig-sekvensen genom att ansluta k-Mer-banan, vilket resulterade i en contig N50 2,104 bp och total längd 2,295,545,898 bp. (2) byggnadsställning: vi fick 80% av alla inriktade Parade slutläsningar genom att justera alla användbara läsningar på contigs. Sedan beräknade vi mängden delade Parade slutförhållanden mellan varje par av contigs, vägde hastigheten för konsekventa och motstridiga Parade ändar och konstruerade sedan ställningarna steg för steg. Som ett resultat fick vi byggnadsställningar med en N50 7,168,038 bp och total längd 2,355,303,269 bp från korta insatsstora Parade ändar till långa avlägsna Parade ändar. (3) Gap closing: för att fylla luckorna inuti de konstruerade ställningarna använde vi den parade informationen för att hämta läsparen för att göra en lokal montering igen för dessa insamlade läsningar. Sammanfattningsvis stängde vi 87.7% av mellanrummen inom ställningen, eller 85.8% av summan gap längd. Contig N50-storleken ökade från 2 104 bp till 21 301 bp (tabell 3). Byggnadsställningsstorleken var 2,355,303,269 bp, som ligger nära den monteringsbaserade genomstorleken på 2,374,716,107 bp rapporterad för den randiga hyaena, Hyaena hyaena11 (NCBI-anslutning: ASM300989v1). Vi hämtade också och kommenterade mitokondriella genomet av den prickiga hyenan med hjälp av MitoZ-programet16, som har en längd av 16,858 bp, liknande de första mitokondriella genomen sekvenserade för denna art12.
bedömning av utkastet till genom utfördes genom att titta på fullständigheten av ortologer med en kopia med BUSCO (version 3.1.0)17, söka mot Mammaliaodb9-databasen som innehåller 4 104 ortologgrupper med en kopia. Totalt identifierades 95,5% av orthologerna som fullständiga, 2,5% som fragmenterade och 2,0% som saknade, vilket indikerar en övergripande hög kvalitet på den fläckiga hyenagenomet. Med tanke på att 99,95% av de korta ställningarna (<1k) endast innehöll 1.2% av den totala genomlängden utesluter vi dessa byggnadsställningar för nedströmsanalys, inklusive repetitivt element och genfunktionsanteckning.
repetitiv elementanteckning
både tandemupprepningar och transponerbara element (TE) sökte efter och identifierades över C. crocuta-genomet. Tandemupprepningar identifierades med hjälp av tandemupprepningar Finder (TRF, v4.07)18 och transponerbara element (TEs) identifierades genom en kombination av homologibaserade och de novo-metoder. För den homologibaserade förutsägelsen använde vi RepeatMasker version 4.0.619 med inställningarna ” – nolow-no_is-norna-engine ncbi ”och RepeatProteinMask (ett program inom RepeatMasker-paketet) med inställningarna”- engine ncbi-noLowSimple-pvalue 0.0001 ” för att söka te på nukleotid-och aminosyranivån baserat på kända upprepningar (Fig. 3). RepeatMasker applicerades för DNA-nivåidentifiering med hjälp av ett anpassat bibliotek som kombinerade Repbase21.10 dataset20. På proteinnivån användes Repeatprotein mask för att utföra RMBlast mot te-proteindatabasen. För ab initio förutsägelse, RepeatModeler (v1.0.8)21 och LTR_FINDING (v1.06) 22 användes för att konstruera de novo repeat library. Kontaminering och flera kopieringssekvenser i biblioteket togs bort och de återstående sekvenserna klassificerades enligt SPRÄNGRESULTATET efter anpassning till SwissProt-databasen. Baserat på detta bibliotek använde vi RepeatMasker för att maskera de homologa TEs och klassificerade dem (Fig. 4). Sammantaget identifierades totalt 826 Mb repetitiva element i den fläckiga hyenan, innefattande 35,29% av hela genomet (Tabell 4).

fördelning av divergenshastighet för varje typ av transponerbart element (TE) i Crocuta crocuta genomaggregat baserat på homologibaserad förutsägelse. Divergenshastigheten beräknades mellan de identifierade TEs i genomet med användning av en homologibaserad metod och konsensussekvensen i Repbase database20.

fördelning av divergenshastigheten för varje typ av TE i Crocuta crocuta genomet montering baserat på ab initio förutsägelse. Divergenshastigheten beräknades mellan de identifierade TEs i genomet av ab initio förutsägelse och konsensussekvensen i det förutsagda te-biblioteket (se metoder).
Protein-kodande gen annotation
vi använde ab initio förutsägelse och homolog-baserade metoder för att kommentera protein-kodande gener samt skarvning platser och alternativa skarvning isoformer. Ab initio prediction utfördes på det upprepade maskerade genomet med hjälp av genmodeller från människa, tamhund och tamkatt med AUGUSTUS (version 2.5.5)23, GENSCAN24, GlimmerHMM (version 3.0.4)25 och SNAP (version 2006-07-28)26. Totalt identifierades 22 789 gener med denna metod. Homologa proteiner av, Homo sapiens, Felis catus och Canis familiaris (från Ensembl 96-utgåvan) kartlades till det fläckiga hyengenomet med användning av tblastn (Blastall 2.2.26)27 med parametrar ”-e 1e-5”. De justerade sekvenserna såväl som deras frågeproteiner skickades sedan till GeneWise (version 2.4.1)28 för att söka efter en exakt skarvad inriktning. Den slutliga genuppsättningen (22 747) samlades in genom sammanslagning av ab initio och homologbaserade resultat med hjälp av en anpassad pipeline (Tabell 5).
genfunktionsanteckning
Genfunktioner tilldelades enligt den bästa matchningen som erhölls genom att anpassa översatta genkodningssekvenser med BLASTP med parametrar ”-e 1e-5” till SwissProt-och TrEMBL-databaserna (Uniprot release 2017-09). Genernas motiv och domäner bestämdes av InterProScan (v5)29 mot proteindatabaser inklusive ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 och PROSITE35. Gen ontologi-ID för varje gen erhölls från motsvarande SwissProt-och TrEMBL-poster. Alla gener justerades mot KEGG-proteiner, och vägen i vilken genen kan vara involverad härleddes från de matchade generna i KEGG-databasen36. Sammanfattningsvis kommenterades 22 166 (97,45%) av de förutsagda proteinkodande generna framgångsrikt av minst en av de sex databaserna (Tabell 6).
Genfamiljkonstruktion och fylogeni rekonstruktion
för att få insikt i den fylogenetiska historien och utvecklingen av genfamiljer av Crocuta crocuta, grupperade vi gensekvenser av sju arter (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) och Homo sapiens som utgrupp (Ensembl release-96, Panthera leo från opublicerade data) till genfamiljer som använder Orthomcl (v2.0.9)37. De proteinkodande generna för de åtta arterna hämtades genom att välja den längsta transkriptisoformen för varje gen för nedströms parvis tilldelning (grafbyggnad). Vi utförde en all-against-all BLASTP-sökning på proteinsekvenserna av alla referensarter, med ett e-värde cut-off av 1E-5. Genfamiljkonstruktion använde MCL-algoritmen38 med inflationsparametern ’1.5’. Totalt 16 271 genfamiljer av C. crocuta, H. sapiens, F. catus, A. melanoleuca grupperades. Det fanns 11 671 genfamiljer delade av dessa fyra arter, medan 292 genfamiljer innehållande 1 446 gener var specifika för C. Crocuta (Fig. 5). Märkbart var genfamiljerna C. crocuta och F. catus delade mindre än C. crocuta och H. sapiens delade, vilket kan bero på att H. sapiens hade ett mer fullständigt genom och anteckning.
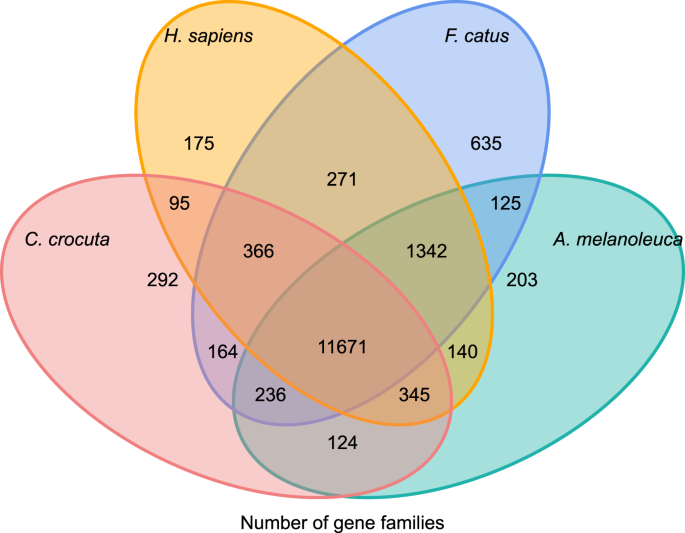
Venn diagram som visar jämförelse av delade och unika proteinkodande gener bland fläckig hyena, människa, huskatt och tamhund baserat på orthology analys.
vi identifierade 6 601 orthologa gener med en kopia för att rekonstruera det fylogenetiska trädet hos de åtta arterna. Flera sekvensinriktningar av aminosyrasekvenser för varje gen genererades med användning av muskler (version 3.8.31)39 och trimmades med Gblock (0.91 b)40, vilket uppnådde väljusterade regioner med parametrarna ”-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”. Vi utförde fylogenetisk analys med maximal sannolikhetsmetod som implementerad i PhyML (v3.0)41, med användning av JTT + G + i-modellen för aminosyrasubstitution (Fig. 6). Trädets rot bestämdes genom att minimera höjden på hela trädet via Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Slutligen uppskattade vi divergenstiden bland de åtta linjerna med MCMCTree från programvarupaketet PAML version 4.442. Två priorer baserade på fossilregistret användes för att kalibrera substitutionsgraden, inklusive Boreoeutheria (91-102 MYA) och Carnivora (52-57 MYA)43. I överensstämmelse med tidigare studier, de prickiga hyenagrupperna med de fyra arterna som ingår från Felidae i en klad som definierar underordningen Feliformia, som avviker från Caniformia (representerad av tamhunden och jättepandaen) 53,9 Mya44.

fylogenetiskt träd av C. crocuta och sju andra arter konstruerade med maximal sannolikhetsmetod baserad på 6 601 enstaka ortologer. Divergenstiden uppskattades med hjälp av de två kalibreringspriorerna härledda från Tidsträddatabasen (http://www.timetree.org), som är markerade med en röd romb. Alla beräknade divergenstider visas med 95% konfidensintervall inom parentes.