monsterverzameling, bibliotheekconstructie en sequencing
genomisch DNA werd verkregen van een mannelijk exemplaar van C. crocuta (NCBI taxonomy ID: 9678; Fig. 1) opgeslagen in de Frozen Zoo® bij het San Diego Zoo Institute for Conservation Research, USA (Frozen Zoo ID: KB4526).
het genomische DNA werd geëxtraheerd met fenolchloroform, gevolgd door zuivering met ethanolprecipitatie13. Het geëxtraheerde DNA werd in werking gesteld en gevisualiseerd op een 1.5% agarose gel in 1x TBE buffer om te controleren op de aanwezigheid van hoog moleculair gewicht DNA. De concentratie en de zuiverheid van DNA werden gekwantificeerd op een NanoDrop 2000 spectrofotometer en Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA) alvorens aan BGI-Shenzhen, China te verschepen. We verkregen een totaal van 372 µg genomisch DNA, met een concentratie van 0,418 µg/µL met behulp van de Nanodrop 2000 en 0,245–0,399 µg/µL op basis van vier replicate lezingen met behulp van de Qubit 2.0 Fluorometer. De 260/280 Verhouding van zuiverheid was 1,95. We barcode vervolgens het monster met behulp van cytochroom B (Cytb) gen. Vervolgens bouwden we volgens de gradiëntbibliotheek-strategie 13 wisselplaatformaatbibliotheken, met de volgende wisselplaatformaatlengtes: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. We hebben de HiSeq gebruikt. 2000 sequencer (Illumina, USA) naar Sequence Paired-End (PE) leest voor elke bibliotheek over 14 rijstroken. Een totaal van ongeveer 299 Gb ruwe gegevens werd gegenereerd uit 13 bibliotheken, het bereiken van een sequencing diepte (dekking) van 149,25 (Tabel 1).
kwaliteitscontrole
om fouten in verkeerde assemblage te minimaliseren, filterden we ruwe reads voorafgaand aan de novo genoomassemblage volgens de volgende twee criteria. Eerst werden reads met meer dan 10 bp uitgelijnd op de adaptersequentie (waarbij <= 3 bp mismatch) verwijderd. Ten tweede werden reads met 40% basen met een kwaliteitswaarde van minder dan of gelijk aan 10 weggegooid. Tenslotte hebben we 190,4 G gegevens verkregen met een dekking van 95,2 (Tabel 2).
schatting van de genoomgrootte
drie short-insert-bibliotheken (twee van 170 bp en één van 500 bp) werden gebruikt om de genoomgrootte en genoombrede heterozygositeit te schatten door middel van k-mer-analyse. Een totaal van ongeveer 385 M PE reads werden ingediend bij jellyfish14 om K-Mer frequentie te berekenen. Vervolgens werd de k-mer distributie geïllustreerd door Genomescope7 met parameters “k = 17; lengte = 100; max coverage = 1000”. We verkregen een geschatte genoomgrootte 2,003,681,234 bp, en heterozygositeit van 0,325% (Fig. 2).

17-Mer schatting van genoomgrootte. De x-as is diepte (X), De y-as is de verhouding die de frequentie bij die diepte vertegenwoordigt gedeeld door de totale frequentie van alle dekkingsdieptes. Zonder rekening te houden met het sequentiefoutpercentage, de heterozygositeitssnelheid en de herhalingssnelheid van het genoom, moet de 17-mer-distributie een Poisson-distributie benaderen.
genome assembly and assessment
SOAPdenovo (V1.06) 15 werd aangewend om het genoom de novo te assembleren, na het filteren van de korte gegevens van de tussenvoegselgrootte en het verwijderen van de kleine piek van de grote gegevens van de tussenvoegselgrootte. De SOAPdenovo assembly algoritme omvatte drie belangrijke stappen. (1) Contig constructie: de short-insert size library data werden opgesplitst in k-mers en geconstrueerd met behulp van een de Bruijn grafiek, die werd vereenvoudigd door het verwijderen van tips, het samenvoegen van bubbels, het verwijderen van de lage dekking van de verbinding en het verwijderen van kleine herhalingen. We verkregen de Contig-sequentie door het k-Mer-pad te verbinden, wat resulteerde in een contig N50 2.104 bp, en totale lengte 2.295.545.898 bp. (2) steigerconstructie: we verkregen 80% van alle uitgelijnde paired-end reads door alle bruikbare lees op contigs opnieuw uit te lijnen. Vervolgens berekenden we het aantal gedeelde gepaarde eindrelaties tussen elk paar contigs, gewogen De snelheid van consistente en conflicterende gepaarde eindpunten, en bouwden de steigers stap voor stap. Als gevolg daarvan verkregen we steigers met een N50 7.168.038 bp, en totale lengte 2,355.303.269 bp van korte insert-sized gepaarde-uiteinden, aan lange verre gepaarde-uiteinden. (3) Gap closing: om de gaten in de geconstrueerde steigers te vullen, gebruikten we de gepaarde-end informatie om de leesparen op te halen om een lokale assemblage opnieuw te doen voor deze verzamelde leest. Samengevat hebben we 87,7% van de intra-scaffold hiaten gesloten, of 85,8% van de sum gap lengte. De grootte van contig N50 steeg van 2.104 bp tot 21.301 bp (Tabel 3). De grootte van de Steigermontage was 2.355.303.269 bp, wat dicht bij de op assemblage gebaseerde genoomgrootte van 2.374.716.107 BP ligt, gerapporteerd voor de gestreepte hyaena, Hyaena hyaena11 (NCBI-toetreding: ASM300989v1). We hebben ook het mitochondriale genoom van de gevlekte hyena opgehaald en geannoteerd met behulp van het MitoZ-programma16, dat een lengte heeft van 16.858 bp, vergelijkbaar met de eerste mitochondriale genomen die voor deze species worden gesequenced12.
beoordeling van het concept genoom werd uitgevoerd door te kijken naar de volledigheid van single-copy orthologs met behulp van BUSCO (Versie 3.1.0)17, zoeken tegen Mammaliaodb9 database die 4.104 single-copy ortholog groepen bevat. Een totaal van 95,5% van de orthologen werd geïdentificeerd als compleet, 2,5% als gefragmenteerd en 2,0% als vermist, wat wijst op een algemene hoge kwaliteit van de gevlekte hyena genoom assemblage. Aangezien 99,95% van de korte steigers (<1k) slechts 1 aanhield.2% van de totale genoomlengte, we uitgesloten deze scaffolds voor downstream analyse, met inbegrip van repetitieve element en Gen functie annotatie.
repetitieve elementannotatie
zowel tandemherhalingen als transposeerbare elementen (te) werden gezocht en geïdentificeerd in het C. crocuta-genoom. Tandem herhalingen werden geà dentificeerd gebruikend Tandem herhalingen Finder (TRF, v4.07)18 en transposable elementen (TEs) werden geà dentificeerd door een combinatie van homologie-gebaseerde en de novo benaderingen. Voor de homologie gebaseerde voorspelling hebben we RepeatMasker versie 4.0 gebruikt.619 met de Instellingen “- nolow-no_is-norna-engine ncbi ” en Repeatproteinmasker (een programma binnen RepeatMasker pakket) met de Instellingen “-engine ncbi-noLowSimple-pvalue 0.0001” om TEs te zoeken op het nucleotide en aminozuur niveau gebaseerd op bekende herhalingen (Fig. 3). RepeatMasker werd toegepast voor DNA-niveau identificatie met behulp van een aangepaste bibliotheek die de Repbase21.10 dataset20 combineerde. Op het eiwitniveau werd het Repeatproteïnmasker gebruikt om RMBlast uit te voeren tegen de te-eiwitdatabase. Voor Ab initio voorspelling, RepeatModeler (v1.0.8)21 en LTR_FINDING (v1.06) 22 werden toegepast om de de novo repeat bibliotheek te bouwen. Contaminatie en Multi-copy sequenties in de bibliotheek werden verwijderd en de resterende sequenties werden geclassificeerd volgens het EXPLOSIERESULTAAT na uitlijning naar de swissprot database. Op basis van deze bibliotheek gebruikten we RepeatMasker om de homologe TEs te maskeren en geclassificeerd (Fig. 4). In totaal werden in de gevlekte hyena in totaal 826 Mb repetitieve elementen geïdentificeerd, waarvan 35,29% van het gehele genoom (Tabel 4).

verdeling van de divergentiesnelheid van elk type transposeerbaar element (te) in de crocuta crocuta genoom assemblage op basis van homologie gebaseerde voorspelling. Het divergentiepercentage werd berekend tussen de geïdentificeerde TEs in het genoom met behulp van een op homologie gebaseerde methode en de consensusvolgorde in de Repbase database20.

verdeling van de divergentiesnelheid van elk type TE in de crocuta crocuta genoom assemblage gebaseerd op Ab initio voorspelling. Het divergentiepercentage werd berekend tussen de geïdentificeerde TEs in het genoom door Ab initio voorspelling en de consensusvolgorde in de voorspelde te bibliotheek (zie methoden).
Eiwitcoderende genannotatie
we gebruikten ab initio voorspelling en homolog-gebaseerde benaderingen om eiwitcoderende genen te annoteren, evenals splicing sites en alternatieve splicing isovormen. Ab initio voorspelling werd uitgevoerd op de repeat-gemaskerde genoom met behulp van Gen modellen van de mens, de hond en de kat in het huis met AUGUSTUS (Versie 2.5.5)23, GENSCAN24, GlimmerHMM (versie 3.0.4)25, en SNAP (versie 2006-07-28)26, respectievelijk. Een totaal van 22.789 genen werden geà dentificeerd door deze methode. Homologe eiwitten van Homo sapiens, Felis catus en Canis familiaris (uit de Ensembl 96 release) werden in kaart gebracht aan het gevlekte hyena genoom met behulp van tblastn (Blastall 2.2.26)27 met parameters “-e 1e-5”. De uitgelijnde opeenvolgingen evenals hun vraagproteã nen werden toen voorgelegd aan GeneWise (Versie 2.4.1)28 voor het zoeken van een nauwkeurige verbonden aanpassing. De uiteindelijke genenset (22.747) werd verzameld door het samenvoegen van Ab initio en homolog-gebaseerde resultaten met behulp van een aangepaste pijplijn (Tabel 5).
genfunctie-annotatie
Genfuncties werden toegewezen volgens de beste overeenkomst die werd verkregen door vertaalde gencodeersequenties met BLASTP uit te lijnen met parameters “-e 1e-5” in de swissprot-en TrEMBL-databases (Uniprot release 2017-09). De motieven en domeinen van genen werden bepaald door InterProScan (v5)29 tegen eiwitdatabases waaronder ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 en PROSITE35. Genontologie ID ‘ s voor elk gen werden verkregen uit de overeenkomstige swissprot en TrEMBL inzendingen. Alle genen werden tegen Kegg-proteã nen afgestemd, en de weg waarin het gen zou kunnen worden betrokken werd afgeleid van de gematchte genen in de Kegg database36. Samengevat werden 22.166 (97,45%) van de voorspelde eiwitcoderende genen met succes geannoteerd door ten minste één van de zes databases (Tabel 6).
Gen familie bouw en fylogenie-reconstructie
om inzicht Te krijgen in de fylogenetische geschiedenis en de evolutie van het gen families van Crocuta crocuta, we geclusterde gen-sequenties van zeven soorten (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) en de Homo sapiens als de outgroup (Ensembl release-96, Panthera leo van ongepubliceerde gegevens) in gen gezinnen met orthoMCL (v2.0.9)37. De eiwit-coderende genen voor de acht species werden teruggehaald door het langste transcript isovorm voor elk gen voor stroomafwaartse paarsgewijze toewijzing (grafiekenbouw) te selecteren. We voerden een all-against-all BLASTP-onderzoek uit op de eiwitsequenties van alle referentiesoorten, met een e-waarde cut-off van 1e-5. De constructie van de genfamilie gebruikte de MCL-algoritm38 met de inflatieparameter van ‘1.5’. Een totaal van 16.271 genfamilies van C. crocuta, H. sapiens, F. catus, A. melanoleuca werden geclusterd. Er waren 11.671 genfamilies gedeeld door deze vier soorten, terwijl 292 genfamilies met 1.446 genen specifiek waren voor C. Crocuta (Fig. 5). Opmerkelijk, waren de genfamilies Die C. crocuta en F. catus deelden minder dan C. crocuta en H. sapiens deelden, wat kon voortvloeien uit dat H. sapiens een vollediger genoom en annotatie had.
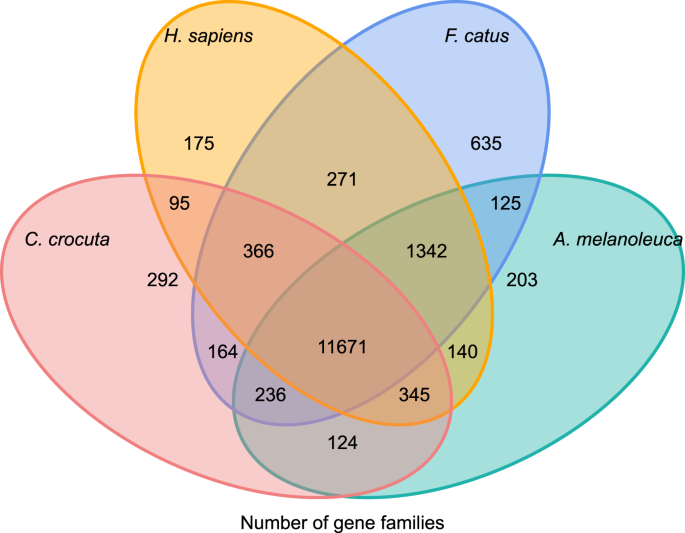
Venn-diagram met een vergelijking van gedeelde en unieke eiwitcoderende genen tussen gevlekte hyena, mens, huiskat en huishond op basis van orthopedische analyse.
we identificeerden 6.601 enkel-kopie orthologe genen om de fylogenetische boom van de acht soorten te reconstrueren. Meerdere sequentie-uitlijningen van aminozuursequenties voor elk gen werden gegenereerd met behulp van spier (versie 3.8.31)39, en getrimd met behulp van Gblocks (0,91 b)40, waardoor goed uitgelijnde gebieden met de parameters “-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”werden bereikt. We voerden fylogenetische analyse uit met behulp van de maximale waarschijnlijkheid methode zoals geïmplementeerd in PhyML (v3.0)41, met behulp van het JTT + G + I model voor aminozuursubstitutie (Fig. 6). De wortel van de boom werd bepaald door het minimaliseren van de hoogte van de hele boom via Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Tot slot hebben we de divergentietijd tussen de acht lijnen geschat met behulp van MCMCTree van het paml versie 4.4 softwarepackage42. Twee priors gebaseerd op het fossielenbestand werden gebruikt om de substitutiesnelheid te kalibreren, waaronder Boreoeutheria (91-102 MYA) en Carnivora (52-57 MYA)43. In overeenstemming met eerdere studies, de gevlekte hyena groepen met de vier soorten opgenomen uit de Felidae in een clade definiëren van de onderorde Feliformia, die afwijken van de Caniformia (vertegenwoordigd door de tamme hond en reuzenpanda) 53,9 Mya44.

Fylogenetic tree of C. crocuta and seven other species construated by the maximum likelihood method based on 6.601 single-copy orthologues. De divergentietijd werd geschat aan de hand van de twee kalibratiepoors die zijn afgeleid van de Tijdboomdatabase (http://www.timetree.org), die zijn gemarkeerd door een rode ruit. Alle geschatte divergentietijden worden weergegeven met 95% betrouwbaarheidsintervallen tussen haakjes.