Sample collection, library construction and sequencing
Genomic DNA was obtained from a male specimen of C. crocuta (NCBI taxonomy ID: 9678; Fig. 1) przechowywany w Frozen Zoo® w San Diego Zoo Institute for Conservation Research, USA (Frozen Zoo ID: KB4526).
genomowy DNA ekstrahowano za pomocą fenolu-chloroformu, a następnie oczyszczano za pomocą wytrącania etanolu13. Pobrane DNA zostało zbadane i zwizualizowane na 1.5% żelu agarozowego uruchamia się w 1X buforze TBE w celu sprawdzenia obecności DNA o dużej masie cząsteczkowej. Stężenie i czystość DNA oznaczono ilościowo na spektrofotometrze NanoDrop 2000 i Fluorometrze Qubit 2.0 (Thermo Fisher Scientific, USA) przed wysyłką do BGI-Shenzhen w Chinach. Otrzymaliśmy łącznie 372 µg genomowego DNA o stężeniu 0,418 µg/µL przy użyciu Nanodrop 2000 i 0,245–0,399 µg / µL na podstawie czterech replikowanych odczytów przy użyciu Fluorometru Qubit 2.0. Stosunek czystości 260/280 wynosił 1,95. Następnie zakodowaliśmy próbkę za pomocą genu cytochromu b (Cytb). Następnie, zgodnie ze strategią biblioteki gradientowej, zbudowaliśmy 13 bibliotek o następujących długościach: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Użyliśmy HiSeq. 2000 sequencer (Illumina, USA) to Sequence Paired-End (PE) czyta dla każdej biblioteki na 14 torach. W sumie około 299 GB surowych danych zostało wygenerowanych z 13 bibliotek, osiągając głębokość sekwencjonowania (pokrycie) 149,25 (Tabela 1).
kontrola jakości
aby zminimalizować błędy w montażu, przefiltrowaliśmy surowe odczyty przed montażem genomu de novo zgodnie z następującymi dwoma kryteriami. Po pierwsze, odczyty z więcej niż 10 bp wyrównane do sekwencji adaptera (pozwalając <= 3 bp niedopasowania) zostały usunięte. Po drugie, odczyty z 40% baz o wartości jakości mniejszej lub równej 10 zostały odrzucone. Ostatecznie uzyskaliśmy dane 190,4 G z pokryciem 95,2 (Tabela 2).
Estymacja rozmiaru genomu
trzy biblioteki z krótkimi wstawkami (dwie z 170 bp i jedna z 500 bp) zostały użyte do oszacowania rozmiaru genomu i heterozygotyczności całego genomu za pomocą analizy k-mer. Łącznie do jellyfish14 przekazano około 385 m odczytów PE w celu obliczenia częstotliwości k-mer. Następnie rozkład k-mer został zilustrowany przez Genomescope7 z parametrami „k = 17; długość = 100; maksymalne pokrycie = 1000”. Otrzymaliśmy szacowaną wielkość genomu 2,003,681,234 bp, a heterozygotyczność 0,325% (rys. 2).

17-Mer oszacowanie wielkości genomu. Oś x jest głębokością (X), oś y jest proporcją, która reprezentuje częstotliwość na tej głębokości podzieloną przez całkowitą częstotliwość wszystkich głębokości pokrycia. Bez uwzględnienia błędu sekwencji, heterozygotyczności i powtarzalności genomu, rozkład 17-mer powinien być zbliżony do rozkładu Poissona.
montaż i ocena genomu
SOAPdenovo (V1.06) 15 został zastosowany do zmontowania genomu de novo, po filtrowaniu krótkich danych o rozmiarze wkładki i usunięciu małego piku danych o dużym rozmiarze wkładki. Algorytm montażu SOAPdenovo obejmował trzy główne etapy. (1) Konstrukcja Contig: dane biblioteki rozmiarów krótkich wstawek zostały podzielone na k-mers i skonstruowane przy użyciu grafu de Bruijna, który został uproszczony przez usunięcie końcówek, scalenie pęcherzyków, usunięcie niskiego pokrycia połączenia i usunięcie małych powtórzeń. Otrzymaliśmy sekwencję contiga łącząc ścieżkę k-mer, w wyniku czego otrzymaliśmy contiga N50 2,104 bp, a Długość całkowita 2,295,545,898 bp. (2) Konstrukcja rusztowania: uzyskaliśmy 80% wszystkich wyrównanych, sparowanych odczytów końcowych, wyrównując wszystkie użyteczne odczyty na stykach. Następnie obliczyliśmy ilość wspólnych parowanych związków końców między każdą parą styków, ważyliśmy szybkość spójnych i sprzecznych parowanych końców, a następnie konstruowaliśmy rusztowania krok po kroku. W rezultacie otrzymaliśmy rusztowania z N50 7,168,038 bp, a całkowita długość 2,355,303,269 bp od krótkich sparowanych końcówek o rozmiarach wkładek do długich odległych sparowanych końców. (3) zamykanie luk: aby wypełnić luki wewnątrz zbudowanych rusztowań, użyliśmy sparowanych informacji końcowych, aby pobrać pary odczytu, aby ponownie wykonać lokalny montaż dla tych zebranych odczytów. Podsumowując, zamknęliśmy 87,7% szczelin wewnątrzzakładowych, czyli 85,8% długości szczeliny sumarycznej. Rozmiar contig N50 wzrósł z 2104 bp do 21301 bp (Tabela 3). Rozmiar zespołu rusztowania wynosił 2,355,303,269 bp, co jest zbliżone do rozmiaru genomu opartego na montażu 2,374,716,107 bp zgłoszonego dla pasiastej hyaena, Hyaena hyaena11 (przystąpienie NCBI: ASM300989v1). Pobraliśmy również i opisaliśmy Genom mitochondrialny hieny plamistej za pomocą programu MitoZ 16, który ma długość 16 858 bp, podobnie jak pierwsze genomy mitochondrialne zsekwencjonowane dla tego gatunku12.
ocena projektu genomu została przeprowadzona poprzez zbadanie kompletności ortologów jednokrotnych przy użyciu BUSCO (Wersja 3.1.0)17, przeszukując bazę danych Mammaliaodb9 zawierającą 4104 jednokrotne grupy ortologów. Łącznie 95,5% ortologów zostało zidentyfikowanych jako kompletne, 2,5% jako fragmentaryczne, a 2,0% jako brakujące, co wskazuje na ogólną wysoką jakość zespołu genomu hieny plamistej. Biorąc pod uwagę, że 99,95% krótkich rusztowań (<1k) miało tylko 1.2% całkowitej długości genomu wykluczyliśmy te rusztowania do dalszej analizy, w tym powtarzalnych elementów i adnotacji cech genów.
adnotacja elementów powtarzalnych
zarówno powtórzenia tandemowe, jak i elementy transponowalne (TE) zostały wyszukane i zidentyfikowane w całym genomie C. crocuta. Powtórzenia tandemowe zidentyfikowano za pomocą narzędzia Tandem Repeats Finder (TRF, v4.07)18, a elementy transponowalne (tes) zidentyfikowano za pomocą kombinacji metod opartych na homologii i de novo. Do przewidywania opartego na homologii użyliśmy RepeatMasker w wersji 4.0.619 z ustawieniami „- nolow-no_is-norna-engine ncbi ” i RepeatProteinMask (program w pakiecie RepeatMasker) z ustawieniami „-engine ncbi-noLowSimple-pvalue 0.0001” do wyszukiwania TEs na poziomie nukleotydów i aminokwasów na podstawie znanych powtórzeń (rys. 3). RepeatMasker został zastosowany do identyfikacji na poziomie DNA przy użyciu niestandardowej biblioteki, która połączyła zestaw danych Repbase21.1020. Na poziomie białka, RepeatProteinMask został użyty do wykonywania RMBlast przeciwko bazie danych białka TE. Dla predykcji ab initio, RepeatModeler (v1.0.8)21 i LTR_FINDING (v1.06) 22 zostały zastosowane do budowy biblioteki powtórzeń de novo. Zanieczyszczenia i sekwencje wielokrotnych kopii w bibliotece zostały usunięte, a pozostałe sekwencje zostały sklasyfikowane zgodnie z wynikiem wybuchu po dopasowaniu do bazy danych SwissProt. W oparciu o tę bibliotekę użyliśmy RepeatMasker do zamaskowania homologicznych TEs i sklasyfikowania ich (rys. 4). Ogółem w hienie plamistej zidentyfikowano 826 Mb powtarzalnych elementów, stanowiących 35,29% całego genomu (Tabela 4).

rozkład współczynnika dywergencji każdego typu elementu transponowalnego (TE) w zespole genomu Crocuta crocuta na podstawie predykcji opartej na homologii. Współczynnik rozbieżności obliczono między zidentyfikowanymi TEs w genomie za pomocą metody opartej na homologii a sekwencją konsensusu w bazie danych Repbase 20.

rozkład stopnia dywergencji każdego typu TE w zespole genomu Crocuta crocuta na podstawie predykcji ab initio. Współczynnik rozbieżności został obliczony między zidentyfikowanymi te w genomie przez predykcję ab initio a sekwencją konsensusu w przewidywanej bibliotece TE (patrz metody).
adnotacja genów kodujących białka
do adnotacji genów kodujących białka, jak również miejsc splicingu i alternatywnych izoform splicingu wykorzystaliśmy predykcję ab initio i podejścia oparte na homologu. Predykcję Ab initio przeprowadzono na genomie maskowanym powtórnie z wykorzystaniem modeli genów pochodzących od człowieka, psa domowego i kota domowego przy użyciu odpowiednio AUGUSTUS (Wersja 2.5.5)23, GENSCAN24, GlimmerHMM (wersja 3.0.4)25 i SNAP (wersja 2006-07-28)26. W sumie zidentyfikowano 22 789 genów tą metodą. Homologiczne białka Homo sapiens, Felis catus i Canis familiaris (z wydania Ensembl 96) zostały zmapowane do genomu hieny plamistej za pomocą tblastn (Blastall 2.2.26)27 o parametrach „-e 1E-5”. Wyrównane sekwencje, jak również ich białka zapytań, zostały następnie przesłane do GeneWise (Wersja 2.4.1)28 w celu wyszukania dokładnego spliced alignment. Ostateczny zestaw genów (22747) zebrano poprzez połączenie wyników ab initio i homologu przy użyciu niestandardowego potoku (Tabela 5).
adnotacja funkcji genu
funkcje genu zostały przypisane zgodnie z najlepszym dopasowaniem uzyskanym przez dopasowanie przetłumaczonych sekwencji kodujących geny za pomocą BLASTP o parametrach „-e 1E-5” do baz danych SwissProt i TrEMBL (wydanie Uniprot 2017-09). Motywy i domeny genów oznaczono za pomocą InterProScan (v5)29 w stosunku do baz danych białek, w tym ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 i PROSITE35. Identyfikatory ontologii genów dla każdego genu uzyskano z odpowiednich wpisów SwissProt i TrEMBL. Wszystkie geny były dopasowane do białek KEGG, a szlak, w którym gen może być zaangażowany, pochodzi z dopasowanych genów w bazie danych KEGG36. Podsumowując, 22 166 (97,45%) przewidywanych genów kodujących białka zostało pomyślnie opatrzonych adnotacją w co najmniej jednej z sześciu baz danych (Tabela 6).
Budowa rodziny genów i rekonstrukcja filogenetyczna
aby uzyskać wgląd w historię filogenetyczną i ewolucję rodzin genów Crocuta crocuta, zgrupowaliśmy sekwencje genów siedmiu gatunków (Felis catus, Canis familiaris, ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) i Homo sapiens jako grupę zewnętrzną (Ensembl release-96, Panthera leo z niepublikowanych danych) w rodziny genów za pomocą orthoMCL (v2.0.9)37. Geny kodujące białka dla ośmiu gatunków pobrano przez wybranie najdłuższej izoformy transkryptu dla każdego genu do przypisania parami (budowanie grafu). Przeprowadziliśmy badanie BLASTP na sekwencjach białek wszystkich gatunków odniesienia, z wartością E odcięcia 1E-5. W budowie rodziny genów zastosowano algorytm MCL38 o parametrze inflacji „1,5”. Łącznie zgrupowano 16 271 rodzin genów C. crocuta, H. sapiens, F. catus, A. melanoleuca. Było 11 671 rodzin genów dzielonych przez te cztery gatunki, podczas gdy 292 rodziny genów zawierające 1446 genów były specyficzne dla C. Crocuta (Fig. 5). Zauważalnie rodziny genów C. crocuta i F. catus były mniejsze niż C. crocuta i H. sapiens, co mogło wynikać z tego, że H. sapiens miał pełniejszy Genom i adnotację.
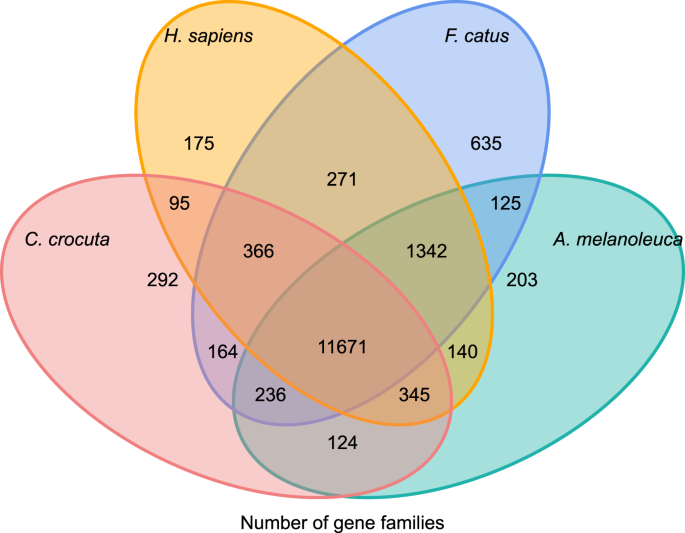
diagram Venna przedstawiający porównanie wspólnych i unikalnych genów kodujących białka wśród hieny plamistej, człowieka, kota domowego i psa domowego na podstawie analizy ortologicznej.
zidentyfikowaliśmy 6601 pojedynczych, ortologicznych genów, aby zrekonstruować drzewo filogenetyczne ośmiu gatunków. Wielokrotne wyrównanie sekwencji aminokwasowych dla każdego genu wygenerowano przy użyciu mięśni (wersja 3.8.31)39 i przycięto przy użyciu Gbloków (0.91 b)40, uzyskując dobrze wyrównane regiony o parametrach „-t = p-b3 = 8-b4 = 10-b5 = n-E = – st”. Wykonaliśmy analizę filogenetyczną metodą maksymalnego prawdopodobieństwa zaimplementowaną w PhyML (v3. 0)41, wykorzystując model JTT + G + I do podstawienia aminokwasów (Fig. 6). Korzeń drzewa został określony przez zminimalizowanie wysokości całego drzewa za pomocą Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Na koniec oszacowaliśmy czas rozbieżności pomiędzy ośmioma liniami wykorzystującymi MCMCTree z pakietu oprogramowania PAML w wersji 4.442. Do skalibrowania stopy substytucji wykorzystano dwa Priory oparte na zapisie kopalnym, w tym Boreoeutheria (91-102 mln lat temu) i Carnivora (52-57 mln lat temu)43. Zgodnie z wcześniejszymi badaniami, hieny plamiste grupują się z czterema gatunkami włączonymi z Felidae do kladu definiującego Podrząd Feliformia, który odbiegał od Caniformia (reprezentowanego przez psa domowego i pandę olbrzymią) 53,9 Mya44.

drzewo filogenetyczne C. crocuta i siedem innych gatunków skonstruowane metodą maksymalnego prawdopodobieństwa na podstawie 6601 ortologów jednokrotnikowych. Czas rozbieżności oszacowano za pomocą dwóch wzorców kalibracyjnych pochodzących z bazy danych drzewa czasu (http://www.timetree.org), które są oznaczone czerwonym rombem. Wszystkie szacowane czasy rozbieżności są pokazane z 95% przedziałami ufności w nawiasach.