prøveindsamling, bibliotekskonstruktion og sekventering
genomisk DNA blev opnået fra en mandlig prøve af C. crocuta (NCBI taksonomi ID: 9678; Fig. 1) opbevares i den frosne dyrepark i San Diego Dyrepark Institut for Conservation Research, USA (frosset Dyrepark ID: KB4526).
det genomiske DNA blev ekstraheret under anvendelse af phenol-chloroform efterfulgt af oprensning ved anvendelse af ethanoludfældning13. Det ekstraherede DNA blev kørt og visualiseret på en 1.5% agarosegel kører i 1 gange TBE-buffer for at kontrollere tilstedeværelsen af DNA med høj molekylvægt. DNA-koncentration og renhed blev kvantificeret på et nanodrop 2000 spektrofotometer og Kvbit 2.0 Fluorometer (Thermo Fisher Scientific, USA) inden forsendelse til BGI-Shengen, Kina. Vi opnåede i alt 372 liter genomisk DNA med en koncentration på 0,418 liter/liter ved hjælp af Nanodrop 2000 og 0,245–0,399 liter/liter baseret på fire replikatmålinger ved hjælp af Kvbit 2,0 Fluorometer. Forholdet 260/280 af renhed var 1,95. Vi barkodede derefter prøven ved hjælp af cytochrom B (Cytb) gen. Derefter konstruerede vi i henhold til gradientbiblioteksstrategien 13 indsatsstørrelsesbiblioteker med følgende indsatsstørrelseslængder: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 KBP. Vi brugte Hisek. 2000 sekvensator (Illumina, USA) til sekvens parret ende (PE) læser for hvert bibliotek på tværs af 14 baner. 299 Gb rådata blev genereret fra 13 biblioteker, hvilket opnåede en sekventeringsdybde (dækning) på 149,25 (tabel 1).
kvalitetskontrol
for at minimere fejl i samlingen filtrerede vi rå aflæsninger inden de novo genomsamling i henhold til følgende to kriterier. Først læser med mere end 10 bp justeret til adaptersekvensen (tillader <= 3 bp mismatch) blev fjernet. For det andet blev læser med 40% af baser med en kvalitetsværdi mindre end eller lig med 10 kasseret. Endelig opnåede vi 190.4 G data med en dækning på 95.2 (tabel 2).
estimering af genomstørrelse
tre kortindsatsbiblioteker (to af 170 bp og en af 500 bp) blev brugt til at estimere genomstørrelsen og genom-bred heterosygositet ved k-mer-analyse. 385 M PE-aflæsninger blev sendt til jellyfish14 for at beregne k-mer-frekvensen. Derefter blev K-mer-fordelingen illustreret af Genomescope7 med parametre “k = 17; længde = 100; maksimal dækning = 1000”. Vi opnåede en estimeret genomstørrelse på 2.003.681.234 bp og heterosygositet på 0,325% (Fig. 2).

17-mer skøn over genom størrelse. Y-aksen er den andel, der repræsenterer frekvensen ved den dybde divideret med den samlede frekvens af alle dækningsdybder. Uden hensyntagen til sekvensfejlfrekvensen, heterosygositetshastigheden og gentagelseshastigheden af genomet, bør 17-mer-fordelingen tilnærme sig en Poisson-fordeling.
genomsamling og vurdering
SOAPdenovo (V1.06) 15 blev anvendt til at samle genomet de novo efter filtrering af de korte indsatsstørrelsesdata og fjernelse af den lille top af de store indsatsstørrelsesdata. SOAPdenovo-samlingsalgoritmen omfattede tre hovedtrin. (1) Contig konstruktion: biblioteksdataene med kort indsats blev opdelt i k-mers og konstrueret ved hjælp af en de Bruijn-graf, som blev forenklet ved at fjerne tip, flette bobler, fjerne den lave dækning af forbindelsen og fjerne små gentagelser. Vi opnåede contig-sekvensen ved at forbinde k-mer-stien, hvilket resulterede i en contig N50 2.104 bp og total længde 2.295.545.898 bp. (2) Stilladskonstruktion: vi opnåede 80% af alle justerede parrede endelæsninger ved at tilpasse alle anvendelige aflæsninger på contigs. Derefter beregnede vi mængden af delte parrede forhold mellem hvert par contigs, vægtede hastigheden af konsistente og modstridende parrede ender, og konstruerede derefter stilladserne trin for trin. Som et resultat opnåede vi stilladser med en N50 7.168.038 bp og total længde 2.355.303.269 bp fra korte indsatsstørrede parrede ender til lange fjerne parrede ender. (3) Gap closing: for at udfylde hullerne inde i de konstruerede stilladser brugte vi de parrede slutoplysninger til at hente læseparene til at lave en lokal samling igen for disse indsamlede læsninger. Sammenfattende lukkede vi 87,7% af hullerne inden for stilladset eller 85,8% af summen af afstanden. Contig N50-størrelsen steg fra 2.104 bp til 21.301 bp (tabel 3). Stilladsstørrelsen var 2.355.303.269 bp, hvilket er tæt på den samlingsbaserede genomstørrelse på 2.374.716.107 BP rapporteret for den stribede hyaena, Hyaena hyaena11 (NCBI-tiltrædelse: ASM300989v1). Vi hentede og kommenterede også mitokondriegenomet af den plettede hyena ved hjælp af mitoseprogrammet16, som har en længde på 16.858 bp, svarende til de første mitokondrielle genomer sekventeret for denne art12.
vurdering af udkastet til genom blev udført ved at se på fuldstændigheden af enkeltkopierede ortologer ved hjælp af BUSCO (version 3.1.0)17 og søge mod Mammaliaodb9-database, der indeholder 4.104 enkeltkopierede ortologgrupper. I alt 95,5% af ortologerne blev identificeret som komplette, 2,5% som fragmenterede og 2,0% som manglende, hvilket indikerer en samlet høj kvalitet af den plettede hyenegenomsamling. I betragtning af at 99,95% af de korte stilladser (<1k) kun havde 1.2% af den samlede genomlængde udelukkede vi disse stilladser til nedstrømsanalyse, herunder gentagne element-og genfunktionsanmærkninger.
Repetitive element annotation
både tandem gentagelser og transponerbare elementer (TE) blev søgt efter og identificeret på tværs af C. crocuta genom. Tandem gentagelser blev identificeret ved hjælp af Tandem Repeats Finder (TRF, v4.07)18 og transponerbare elementer (TEs) blev identificeret ved en kombination af homologi-baserede og de novo tilgange. Til den homologibaserede forudsigelse brugte vi RepeatMasker version 4.0.619 med indstillingerne “- nolov-no_is-norna-engine ncbi” og RepeatProteinMask (et program inden for RepeatMasker-pakken) med indstillingerne “- engine ncbi-nolovsimple-pvalue 0.0001” for at søge TEs på nukleotid-og aminosyreniveauet baseret på kendte gentagelser (Fig. 3). RepeatMasker blev ansøgt om DNA-niveau identifikation ved hjælp af et brugerdefineret bibliotek, der kombinerede Repbase21.10 datasæt20. På proteinniveauet blev RepeatProteinMask brugt til at udføre RMBlast mod TE-proteindatabasen. For ab initio forudsigelse, RepeatModeler (v1.0.8)21 og LTR_FINDING (v1.06) 22 blev anvendt til at konstruere de novo-gentagelsesbiblioteket. Forurening og multi-copy-sekvenser i biblioteket blev fjernet, og de resterende sekvenser blev klassificeret i henhold til EKSPLOSIONSRESULTATET efter justering til databasen. Baseret på dette bibliotek brugte vi RepeatMasker til at maskere de homologe TEs og klassificerede dem (Fig. 4). Samlet set blev i alt 826 Mb gentagne elementer identificeret i den plettede hyena, der omfattede 35,29% af hele genomet (Tabel 4).

fordeling af divergenshastighed for hver type transponerbart element (TE) i Crocuta crocuta genomsamling baseret på homologibaseret forudsigelse. Divergenshastigheden blev beregnet mellem de identificerede TEs i genomet ved hjælp af en homologibaseret metode og konsensussekvensen i Repbase database20.

fordeling af divergenshastighed for hver type TE i Crocuta crocuta genomsamling baseret på Ab initio forudsigelse. Divergenshastigheden blev beregnet mellem de identificerede TEs i genomet ved ab initio forudsigelse og konsensussekvensen i det forudsagte TE-Bibliotek (se metoder).
proteinkodende genanmærkning
vi brugte ab initio-forudsigelse og homolog-baserede tilgange til at kommentere proteinkodende gener såvel som splejsningssteder og alternative splejsningsisoformer. Ab initio forudsigelse blev udført på det gentagne maskerede genom ved hjælp af genmodeller fra human, husdyr og huskat ved hjælp af AUGUSTUS (version 2.5.5)23, GENSCAN24, GlimmerHMM (version 3.0.4)25 og SNAP (version 2006-07-28)26, henholdsvis. I alt 22.789 gener blev identificeret ved denne metode. Homologe proteiner fra, Homo sapiens, Felis catus og Canis familiaris (fra Ensembl 96-udgivelsen) blev kortlagt til det plettede hyenegenom ved hjælp af tblastn (Blastall 2.2.26)27 med parametre “-e 1e-5”. De justerede sekvenser såvel som deres forespørgselsproteiner blev derefter sendt til Genvis (version 2.4.1)28 for at søge en nøjagtig splejset justering. Det endelige gensæt (22.747) blev indsamlet ved at flette ab initio og homolog-baserede resultater ved hjælp af en tilpasset pipeline (tabel 5).
genfunktionsnotation
Genfunktioner blev tildelt i henhold til det bedste match opnået ved at tilpasse oversatte genkodende sekvenser ved hjælp af BLASTP med parametre “-e 1e-5” til databaserne “- e 1e-5 ” (Uniprot release 2017-09). Genernes motiver og domæner blev bestemt af InterProScan (v5) 29 mod proteindatabaser, herunder ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 og PROSITE35. Gen – ontologi-id ‘ er for hvert gen blev opnået fra de tilsvarende Svingprot-og TrEMBL-poster. Alle gener blev justeret mod KEGG-proteiner, og den vej, hvor genet kunne være involveret, blev afledt af de matchede gener i KEGG-databasen36. Sammenfattende blev 22.166 (97,45%) af de forudsagte proteinkodende gener med succes kommenteret af mindst en af de seks databaser (Tabel 6).
Genfamiliekonstruktion og fylogenirekonstruktion
for at få indsigt i den fylogenetiske historie og udvikling af genfamilier af Crocuta crocuta, klyngede vi gensekvenser af syv arter (Felis catus, Canis familiaris, ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) og Homo sapiens som udgruppen (Ensembl release-96, Panthera leo fra upublicerede data) til genet familier, der bruger orthomcl (v2.0.9)37. De proteinkodende gener for de otte arter blev hentet ved at vælge den længste transkriptisoform for hvert gen til nedstrøms parvis tildeling (grafbygning). Vi udførte en all-against-all BLASTP-søgning på proteinsekvenserne af alle referencearten, med en e-værdi cut-off på 1e-5. Gene family construction anvendte MCL algorithm38 med inflationsparameteren ‘1.5’. I alt 16.271 genfamilier af C. crocuta, H. sapiens, F. catus, A. melanoleuca blev grupperet. Der var 11.671 genfamilier delt af disse fire arter, mens 292 genfamilier indeholdende 1.446 gener var specifikke for C. Crocuta (Fig. 5). Mærkbart var genfamilierne C. crocuta og F. catus delte mindre end C. crocuta og H. sapiens delte, hvilket kunne skyldes, at H. sapiens havde et mere komplet genom og annotation.
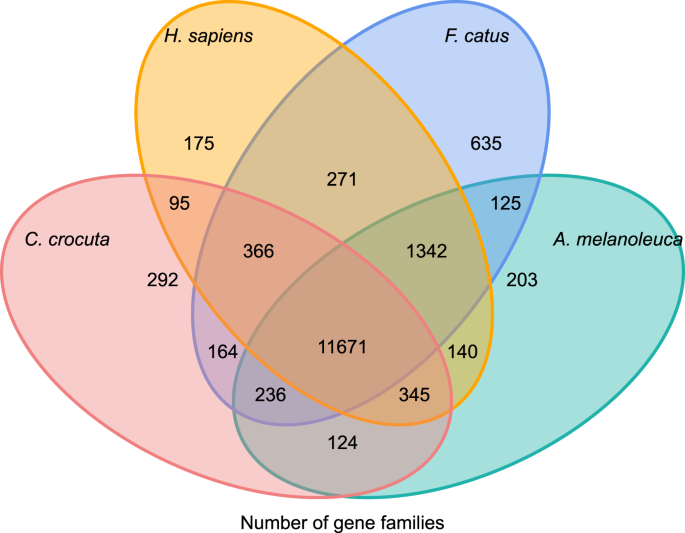
Venn-diagram, der viser sammenligning af delte og unikke proteinkodende gener blandt plettet hyena, menneske, huskat og husdyr baseret på ortologianalyse.
vi identificerede 6.601 ortologe gener med en kopi for at rekonstruere det fylogenetiske træ af de otte arter. Flere sekvensjusteringer af aminosyresekvenser for hvert gen blev genereret ved hjælp af muskel (version 3.8.31)39 og trimmet ved hjælp af Gblocks (0.91 b)40, hvilket opnåede veljusterede regioner med parametrene “-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”. Vi udførte fylogenetisk analyse ved hjælp af metoden med maksimal sandsynlighed som implementeret i PhyML (v3.0)41 Ved hjælp af JTT + G + i-modellen til aminosyresubstitution (Fig. 6). Træets rod blev bestemt ved at minimere højden af hele træet via Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Endelig estimerede vi divergenstiden mellem de otte Slægter ved hjælp af MCMCTree fra PAML version 4.4 programpakke42. To priors baseret på den fossile rekord blev brugt til at kalibrere substitutionshastigheden, herunder Boreeutheria (91-102 MYA) og Carnivora (52-57 MYA)43. I overensstemmelse med tidligere undersøgelser omfattede de plettede hyenagrupper med de fire arter fra Felidae i en klade, der definerede underordenen Feliformia, som divergerede fra Caniformia (repræsenteret af husdyr og kæmpe panda) 53.9 Mya44.

fylogenetisk træ af C. crocuta og syv andre arter konstrueret ved den maksimale sandsynlighedsmetode baseret på 6.601 enkeltkopierede ortologer. Divergenstiden blev estimeret ved hjælp af de to kalibreringspriorer afledt af Tidstrædatabasen (http://www.timetree.org), som er markeret med en rød rombe. Alle estimerede divergenstider vises med 95% konfidensintervaller i parentes.