mintagyűjtés, könyvtárépítés és szekvenálás
a genomi DNS-t a C. crocuta hím példányából nyertük (NCBI taxonomy ID: 9678; ábra. 1) a fagyasztott állatkertben tárolják a San Diego Zoo természetvédelmi Kutatóintézet, USA (fagyasztott Állatkert azonosító: KB4526).
a genomi DNS-t fenol-kloroform alkalmazásával extraháltuk, majd etanol kicsapódással tisztítottuk13. A kivont DNS-t lefuttattuk és vizualizáltuk egy 1-esen.5% agaróz gél fut 1x TBE pufferben a nagy molekulatömegű DNS jelenlétének ellenőrzésére. A DNS koncentrációját és tisztaságát nanodrop 2000 spektrofotométeren és Qubit 2.0 Fluorométeren (Thermo Fisher Scientific, USA) számszerűsítették, mielőtt a BGI-Shenzhen, Kína. Kaptunk összesen 372 µg genomiális DNS-t, a koncentráció, a 0.418 µg/µL segítségével a Nanodrop 2000 0.245–0.399 µg/µL alapján négy párhuzamos mérés segítségével a Qubit 2.0 Fluorometer. A tisztaság 260/280 aránya 1,95 volt. Ezután vonalkódoltuk a mintát citokróm b (Cytb) gén segítségével. Ezután a gradiens könyvtárstratégia szerint 13 betétméretű könyvtárat készítettünk, a következő betétméret-hosszúságokkal: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. A HiSeq-t használtuk. 2000 szekvenszer (Illumina, USA) a szekvencia párosított-End (PE) olvas minden könyvtár egész 14 sávot. Összesen körülbelül 299 Gb nyers adatot generáltak 13 könyvtárból, 149,25 szekvenálási mélységet (lefedettséget) elérve (1.táblázat).
minőségellenőrzés
a hibás összeszerelési hibák minimalizálása érdekében a nyers olvasmányokat a De novo Genom összeszerelése előtt szűrtük a következő két kritérium szerint. Először az adapter sorrendjéhez igazított 10 bp-nél nagyobb leolvasásokat (lehetővé téve <= 3 bp eltérést) eltávolítottuk. Másodszor, a bázisok 40% – ának 10-nél kisebb vagy egyenlő minőségi értékű olvasását elvetettük. Végül 190,4 G adatot kaptunk 95,2 lefedettséggel (2.táblázat).
a genom méretének becslése
három rövid beillesztésű könyvtárat (kettő 170 bp-ből és egy 500 bp-ből) használtunk a genom méretének és a genom egészére kiterjedő heterozigozitásnak a K-mer elemzéssel történő becsléséhez. Összesen körülbelül 385 M PE olvasást nyújtottak be a jellyfish14-hez a k-mer frekvencia kiszámításához. Ezután a K-mer eloszlást a Genomescope7 szemlélteti “k = 17; Hossz = 100; maximális lefedettség = 1000″paraméterekkel. A becsült genomméret 2 003 681 234 bp, a heterozigozitás pedig 0,325% (ábra. 2).

17-a genom méretének mer becslése. Az x tengely mélység (X), az y tengely az az arány, amely az adott mélységben lévő frekvenciát elosztja az összes lefedettségi mélység Teljes frekvenciájával. A szekvencia hibaarány, a heterozigozitás és a genom ismétlési sebességének figyelembevétele nélkül a 17-mer eloszlásnak közelítenie kell a Poisson-eloszlást.
Genom assembly and assessment
SOAPdenovo (V1.06) 15-öt alkalmaztunk a Genome de novo összeállításához, a rövid beszúrási méretadatok szűrését és a nagy beszúrási méretadatok kis csúcsának eltávolítását követően. A SOAPdenovo összeszerelési algoritmus három fő lépést tartalmazott. (1) Contig felépítés: a rövid beszúrható méretű könyvtári adatokat k-mers-re osztottuk, és egy de Bruijn gráf segítségével készítettük el, amelyet egyszerűsítettünk a tippek eltávolításával, a buborékok egyesítésével, a kapcsolat alacsony lefedettségének eltávolításával és a kis ismétlések eltávolításával. A contig szekvenciát a k-mer útvonal összekapcsolásával kaptuk meg, amelynek eredményeként a contig N50 2,104 bp, a teljes hossz pedig 2,295,545,898 bp. (2) Állványszerkezet: az összes igazított párosított végolvasás 80%-át úgy kaptuk meg, hogy az összes használható olvasást átcsoportosítottuk a csatlakozókon. Ezután kiszámítottuk a párosított végek közötti megosztott kapcsolatok mennyiségét, súlyoztuk a konzisztens és ütköző párosított végek arányát, majd lépésről lépésre felépítettük az állványokat. Ennek eredményeként N50 7 168 038 bp-s állványzatokat kaptunk, teljes hossza pedig 2 355 303 269 bp volt a rövid lapka méretű párosított végektől a hosszú, távoli párosított végekig. (3) Részárás: az épített állványokon belüli rések kitöltéséhez a párosított véginformációkat használtuk az olvasási Párok lekérésére, hogy újra helyi összeszerelést végezzünk ezekre az összegyűjtött olvasásokra. Összefoglalva, az állványon belüli rések 87,7%-át, vagyis az összegrés hosszának 85,8% – át zártuk le. A contig N50 mérete 2104 bp-ről 21 301 bp-re nőtt (3.táblázat). Az állványegység mérete 2 355 303 269 bp volt, ami közel áll a csíkos hyaena, Hyaena hyaena11 (NCBI csatlakozás: ASM300989v1) esetében jelentett 2 374 716 107 BP összeszerelési alapú genommérethez. A mitoz program16 segítségével a foltos hiéna mitokondriális genomját is lekértük és megjegyeztük, amelynek hossza 16 858 bp, hasonlóan az e fajhoz szekvenált első mitokondriális genomokhoz12.
a genomvázlat értékelését az egypéldányos ortológusok teljességének vizsgálatával végeztük BUSCO (3.1.0 verzió)17, a Mammaliaodb9 adatbázis alapján, amely 4104 egypéldányos ortológ csoportot tartalmaz. Az ortológusok 95,5% – át teljesnek, 2,5% – át fragmentáltnak, 2,0% – át pedig hiányzónak találták, ami a foltos hiéna Genom összeállításának általános magas színvonalát jelzi. Tekintettel arra, hogy a rövid állványok 99,95% – a (<1k) csak 1-et tartalmazott.A teljes genomhossz 2% – ában kizártuk ezeket az állványokat a downstream elemzéshez, beleértve az ismétlődő elem és a génjellemzők megjegyzését.
ismétlődő elem annotáció
mind a tandem ismétlődéseket, mind a transzponálható elemeket (te) kerestük és azonosítottuk a C. crocuta genomjában. A Tandem ismétléseket a Tandem ismétlések keresőjével (TRF, v4.07)18 azonosítottuk, a transzponálható elemeket (TEs) pedig homológiaalapú és De novo megközelítések kombinációjával azonosítottuk. A homológia alapú előrejelzéshez a RepeatMasker 4.0 verziót használtuk.619 a Beállítások “-nolow-no_is-norna-motor ncbi” és RepeatProteinMask (program belül RepeatMasker csomag) a Beállítások “- motor ncbi-noLowSimple-pfalue 0.0001″keresni TEs a nukleotid és aminosav szinten alapuló ismert ismétlések (ábra. 3). A RepeatMasker-t DNS-szintű azonosításra alkalmazták egy egyedi könyvtár segítségével, amely egyesítette a Repbase21.10 adatkészletet20. Fehérje szinten Repeatproteinmaskot használtunk az RMBLAST végrehajtására a TE fehérje adatbázis ellen. Az ab initio előrejelzéshez, RepeatModeler (v1.0.8)21 és LTR_FINDING (v1.06)22-et alkalmaztak a De novo ismétlés könyvtár. A könyvtárban lévő szennyeződéseket és többmásolatú szekvenciákat eltávolítottuk, a fennmaradó szekvenciákat pedig a robbanás eredménye szerint osztályoztuk a SwissProt adatbázishoz való igazítást követően. Ennek alapján a könyvtár, használtuk RepeatMasker elfedni a homológ TEs és osztályozni őket (ábra. 4). Összességében összesen 826 Mb ismétlődő elemet azonosítottak a foltos hiénában, amely a teljes genom 35,29% – át teszi ki (4.táblázat).

az egyes transzponálható elemek (TE) divergencia sebességének eloszlása a Crocuta crocuta Genom összeállításban homológia alapú előrejelzés alapján. A divergencia arányt a genomban azonosított TEs-ek és a repbase database20 konszenzusos szekvenciája között számítottuk ki homológián alapuló módszerrel.

az egyes TE-típusok divergencia arányának eloszlása a Crocuta crocuta Genom összeállításban ab initio előrejelzés alapján. A divergencia arányt a genomban azonosított TEs-ek között ab initio predikcióval és az előrejelzett te könyvtárban lévő konszenzus szekvenciával számítottuk ki (lásd módszerek).
fehérje kódoló gén annotáció
ab initio predikciót és homológ alapú megközelítéseket használtunk a fehérje kódoló gének, valamint a splicing helyek és az alternatív splicing izoformák annotálására. Az Ab initio predikciót az ismételt maszkos genomon hajtottuk végre az AUGUSTUS (2.5.5 verzió)23, A GENSCAN24, a GlimmerHMM (3.0.4 verzió)25 és a SNAP (2006-07-28 verzió)26 használatával. Összesen 22 789 gént azonosítottak ezzel a módszerrel. A Homo sapiens, a Felis catus és a Canis familiaris homológ fehérjéit (az Ensembl 96 kiadásból) a tblastn (Blastall 2.2.26)27 alkalmazásával, “- e 1E-5″paraméterekkel térképeztük fel a foltos hiéna genomjára. Az igazított szekvenciákat, valamint lekérdező fehérjéiket ezután elküldtük a GeneWise (2.4.1 verzió)28-nak, hogy pontos illesztett igazítást keressenek. A végső génkészletet (22 747) az ab initio és a homolog alapú eredmények összevonásával gyűjtöttük össze egy testreszabott csővezeték segítségével (5.táblázat).
Génfunkció annotáció
a Génfunkciókat a legjobb egyezésnek megfelelően rendeltük hozzá, amelyet a lefordított génkódoló szekvenciák blastp-vel történő “-e 1E-5” paraméterekkel történő összehangolásával nyertünk a SwissProt és a TrEMBL adatbázisokhoz (Uniprot release 2017-09). A gének motívumait és doménjeit az InterProScan (v5) 29 határozta meg a ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 és PROSITE35 protein adatbázisok alapján. Az egyes gének gén ontológiai azonosítóit a megfelelő SwissProt és TrEMBL bejegyzésekből nyertük. Minden gén igazodott a Kegg fehérjékhez, és az útvonal, amelyben a gén részt vehet, a Kegg adatbázisban36 található egyező génekből származik. Összefoglalva, az előrejelzett fehérje-kódoló gének 22 166 (97,45%) sikeresen feljegyezték a hat adatbázis legalább egyikét (6.táblázat).
ahhoz, hogy betekintést nyerjünk a crocuta crocuta géncsaládok filogenetikai történetébe és evolúciójába, hét faj (Felis catus, Canis familiaris, ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) és Homo sapiens génszekvenciáit csoportosítottuk (Ensembl release-96, Panthera leo a nem publikált adatokból) géncsaládokba orthomcl (v2.0.9)37. A nyolc faj fehérjét kódoló génjeit úgy nyertük ki, hogy kiválasztottuk az egyes gének leghosszabb transzkriptum izoformáját a downstream páronkénti hozzárendeléshez (grafikonépítés). Minden ellen blastp keresést végeztünk az összes referencia faj fehérjeszekvenciáján, 1e-5 e-értékhatárral. A géncsalád felépítése az MCL algoritmust alkalmazta38 ‘ 1,5 ‘ inflációs paraméterrel. Összesen 16 271 C. crocuta, H. sapiens, F. catus, A. melanoleuca géncsaládot csoportosítottak. 11 671 géncsaládot osztott meg ez a négy faj, míg 292 géncsalád 1446 gént tartalmazott, amelyek specifikusak voltak a C. Crocuta-ra (ábra. 5). Figyelemre méltó, hogy a C. crocuta és az F. catus közös géncsaládjai kisebbek voltak, mint a C. crocuta és a H. sapiens közös, ami abból eredhet, hogy a H. sapiens teljesebb genommal és annotációval rendelkezett.
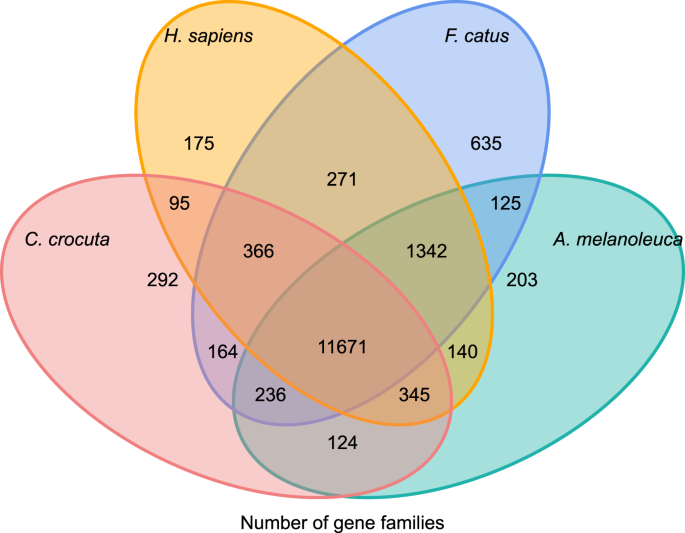
Venn-diagram, amely a megosztott és egyedi fehérjét kódoló gének összehasonlítását mutatja a foltos hiéna, az ember, a házimacska és a házi kutya között ortológiai elemzés alapján.
6601 egypéldányos ortológ gént azonosítottunk a nyolc faj filogenetikai fájának rekonstruálására. Az egyes gének aminosav-szekvenciáinak több szekvenciaillesztését az izom (3.8.31 verzió) 39 segítségével állítottuk elő, és a Gblocks (0.91 b)40 segítségével vágtuk le, jól illeszkedő régiókat érve el a “-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”paraméterekkel. Filogenetikai analízist végeztünk a phyml (v3.0)41-ben megvalósított maximális valószínűségű módszerrel, a JTT + G + i modell alkalmazásával aminosav szubsztitúció (ábra. 6). A fa gyökerét úgy határoztuk meg, hogy minimalizáljuk az egész fa magasságát a Treebesten keresztül (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Végül megbecsültük a nyolc vonal közötti divergencia idejét az MCMCTree segítségével a PAML 4.4-es verziójú szoftvercsomagból42. A helyettesítési Arány kalibrálásához két, a fosszilis rekordokon alapuló priort használtak, köztük a Boreoutheria (91-102 MYA) és a Carnivora (52-57 MYA)43. A korábbi tanulmányokkal összhangban a foltos hiéna a Felidae-ből származó négy fajjal egy olyan kládba csoportosul, amely meghatározza a Feliformia alrendet, amely eltér a Caniformia-tól (amelyet a házi kutya és az óriáspanda képvisel) 53.9 Mya44.

a C. crocuta és hét másik faj filogenetikai fája a maximum likelihood módszerrel készült, 6601 egypéldányos ortológuson alapulva. A divergencia idejét a Time Tree adatbázisból (http://www.timetree.org) származó két kalibrációs prior segítségével becsülték meg, amelyeket vörös rombusz jelöl. Az összes becsült divergencia-időt zárójelben 95% – os konfidencia-intervallummal mutatjuk be.