näytekokoelma, kirjaston rakenne ja sekvensointi
genomin DNA saatiin C. crocutan urosnäytteestä (NCBI taksonomy ID: 9678; Kuva. 1) säilytetään Frozen Zoo®: ssä San Diego Zoo Institute for Conservation Research, USA: ssa (Frozen Zoo ID: KB4526).
genominen DNA uutettiin fenolikloroformilla, minkä jälkeen se puhdistettiin etanolisaostumalla13. Uutettu DNA ajettiin ja visualisoitiin 1.5% agaroosigeeliä käytetään 1x TBE-puskurissa suurimolekyylisen DNA: n toteamiseksi. DNA: n pitoisuus ja puhtaus määritettiin NanoDrop 2000-spektrofotometrillä ja Qubit 2.0-Fluorometrillä (Thermo Fisher Scientific, USA) ennen toimitusta BGI-Shenzheniin Kiinaan. Saimme yhteensä 372 µg genomista DNA: ta, jonka pitoisuus oli 0,418 µg/µL Nanodrop 2000: lla ja 0,245–0,399 µg/µL perustuen neljään toistolukemaan Qubit 2,0-Fluorometrillä. Puhtaussuhde 260/280 oli 1,95. Sitten viivakoodasimme näytteen sytokromi b: n (Sytb) geenillä. Sitten gradienttikirjastostrategian mukaisesti rakensimme 13 inserttikokoista kirjastoa, joissa on seuraavat insert-koon pituudet: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Käytimme Hiseqiä. 2000 sekvensseri (Illumina, USA) sekvenssin pariksi-End (PE) lukee kunkin kirjaston poikki 14 kaistaa. Yhteensä 13 kirjastosta tuotettiin noin 299 Gt raakadataa, jolloin sekvensointisyvyys (kattavuus) oli 149,25 (Taulukko 1).
laadunvalvonta
väärien kokoamisvirheiden minimoimiseksi suodatimme raakalukemat ennen de novo genomikokoonpanoa seuraavien kahden kriteerin mukaisesti. Ensimmäinen, lukee yli 10 bp linjassa sovittimen sekvenssi (mahdollistaa <= 3 bp epäsuhta) poistettiin. Toiseksi, lukee 40% emäkset, joiden laatuarvo on pienempi tai yhtä suuri kuin 10 hylättiin. Lopuksi saimme 190,4 G: n tiedot, joiden kattavuus on 95,2 (Taulukko 2).
genomin kokoa
arvioitiin k-mer-analyysillä kolmella lyhytsisäisellä kirjastolla (kaksi 170 bp ja yksi 500 bp). Jellyfish14: lle lähetettiin yhteensä noin 385 M PE-lukua k-mer-taajuuden laskemiseksi. Sitten k-mer-jakaumaa havainnollisti Genomescope7 parametreilla ”k = 17; length = 100; max coverage = 1000”. Saimme arviolta genomin koon 2,003,681,234 bp, ja heterotsygoottisuus 0,325% (Kuva. 2).

17-mer-arvio genomin koosta. X-akseli on syvyys (X), y-akseli on osuus, joka edustaa taajuutta kyseisellä syvyydellä jaettuna kaikkien peittosyvyyksien kokonaistaajuudella. Ottamatta huomioon genomin sekvenssivirhetasoa, heterotsygoottisuusnopeutta ja toistonopeutta, 17-mer-jakauman pitäisi likimäärin olla Poisson-jakauma.
genomin kokoaminen ja arviointi
SOAPdenovo (V1.06) 15 käytettiin koota genomi de novo, jälkeen suodatus lyhyen insertin koko Tiedot ja poistamalla pieni huippu suuren insertin koko TIEDOT. Soapdenovon kokoonpanoalgoritmi sisälsi kolme päävaihetta. (1) Contig construction: lyhyt-insert koko kirjaston tiedot jaettiin k-mers ja rakennettu käyttäen de Bruijn graafi, jota yksinkertaistettiin poistamalla vinkkejä, yhdistämällä kuplia, poistamalla Alhainen kattavuus yhteyden ja poistamalla pieniä toistoja. Saimme contig-sekvenssin yhdistämällä k-mer-polun, jolloin saatiin contig N50 2,104 bp ja kokonaispituus 2,295,545,898 bp. (2) rakennustelineiden rakentaminen: saimme 80% kaikista kohdistetuista paripäätylukemista uudelleensijoittamalla kaikki käyttökelpoiset lukemat kontigeille. Sitten laskimme määrä jaetun pariksi-end suhteita kunkin parin contigs, painotettu määrä johdonmukainen ja ristiriitaisia pariksi-päättyy, ja sitten rakennettu telineet askel askeleelta. Tämän seurauksena saimme telineet, joissa oli N50 7,168,038 bp, ja kokonaispituus 2,355,303,269 bp lyhyistä insertin kokoisista paripäistä pitkiin kaukaisiin paripuihin. (3) Gap closing: täyttääksemme rakennettujen telineiden sisällä olevat aukot käytimme pariloppuista tietoa noutaaksemme lukuparit tehdäksemme paikalliskokouksen uudelleen näille kerätyille lukemille. Tiivistettynä suljimme 87,7% telineiden sisäisistä raoista eli 85,8% summaerojen pituudesta. Contig N50: n koko kasvoi 2 104 bp: stä 21 301 bp: hen (Taulukko 3). Telineen koko oli 2,355,303,269 bp, mikä on lähellä kokoonpano-pohjainen genomin koko 2,374,716,107 bp raportoitu raidallinen hyaena, Hyaena hyaena11 (NCBI liittyminen: ASM300989v1). Haimme ja kirjasimme myös täplähyeenan mitokondriaalisen genomin mitoz-ohjelmalla16, jonka pituus on 16,858 bp, samanlainen kuin ensimmäiset mitokondriaaliset genomit, jotka sekvensoitiin tälle lajille 12.
Genomiluonnoksen arviointi tehtiin tarkastelemalla yhden kopion ortologien täydellisyyttä BUSCO: n avulla (versio 3.1.0)17, etsimällä Mammaliaodb9-tietokantaa, joka sisältää 4 104 yhden kopion ortologiryhmää. Yhteensä 95,5% ortologeista todettiin täydellisiksi, 2,5% hajanaisiksi ja 2,0% puuttuviksi, mikä viittaa täplähyeenan genomikokoonpanon yleiseen korkeaan laatuun. Ottaen huomioon, että 99,95% lyhyistä telineistä (<1k) kantoi vain 1.2% genomin kokonaispituudesta, jätimme pois nämä telineet loppupään analyysia varten, mukaan lukien toistuva elementti ja geenien ominaisuus merkinnät.
toistuvia alkuaineita koskeva huomautus
sekä tandem-toistoja että transposoituvia alkuaineita (TE) etsittiin ja tunnistettiin C. crocutan genomista. Tandem toistoja tunnistettiin Tandem toistoja Finder (TRF, v4.07)18 ja transposable elements (TEs) tunnistettiin yhdistelmä homology-pohjainen ja de novo lähestymistapoja. Homologiaan perustuvassa ennustuksessa käytimme Toistomaskerin versiota 4.0.619 asetuksilla ” – nolow-no_is-norna-engine ncbi ”ja Toistoproteinmask (ohjelma Toistopaketissa) asetuksilla”- engine ncbi-noLowSimple-pvalue 0.0001 ” etsiä TEs nukleotidi-ja aminohappotasolla tunnettujen toistojen perusteella (Kuva. 3). Repermaskeria haettiin DNA-tason tunnistukseen mukautetulla kirjastolla, joka yhdisti Repbase21.10 dataset20: n. Proteiinitasolla Toistoproteiinimaskia käytettiin Rmblastin suorittamiseen TE-proteiinitietokantaa vastaan. Ab initio prediction, Toistomodeler (v1. 0. 8) 21 ja LTR_FINDING (v1.06) 22 sovellettiin rakentaa de novo toista kirjasto. Kontaminaatio ja monikopiojaksot kirjastossa poistettiin ja loput jaksot luokiteltiin RÄJÄHDYSTULOKSEN mukaan swissprot-tietokantaan kohdistamisen jälkeen. Tämän kirjaston perusteella käytimme Toistomaskeria peittämään homologisia Tesejä ja luokittelimme ne (Kuva. 4). Täplähyeenasta tunnistettiin kaiken kaikkiaan 826 Mb toistuvia alkuaineita, jotka käsittivät 35,29% koko genomista (Taulukko 4).

kunkin transposoituvan alkuaineen (TE) jakauma Crocuta crocuta genomikokoonpanossa homologiaan perustuvan ennusteen perusteella. Genomissa tunnistettujen TEs: ien ja repbase-tietokanta20: n konsensussekvenssin välinen ero laskettiin homologiaan perustuvalla menetelmällä.

kunkin TE-tyypin eroavaisuuksien jakautuminen Crocuta crocuta-genomikokoonpanossa ab initio-ennusteen perusteella. Genomissa Havaittujen TEs: ien ja ennustetun TE-kirjaston konsensusjärjestyksen välinen ero laskettiin ab initio-ennusteella (KS.menetelmät).
proteiinia koodaavan geenin merkintä
käytimme ab initio-ennustus-ja homologipohjaisia lähestymistapoja kirjataksemme proteiinia koodaavia geenejä sekä risteytyspaikkoja ja vaihtoehtoisia risteytysisoformeja. Ab initio ennustus tehtiin toistuvan naamioitu genomi käyttäen geenimalleja ihmisen, koti koira, ja kotikissa käyttäen AUGUSTUS (versio 2.5.5)23, GENSCAN24, GlimmerHMM (versio 3.0.4)25, ja SNAP (versio 2006-07-28) 26, vastaavasti. Tällä menetelmällä tunnistettiin yhteensä 22 789 geeniä. Homologiset proteiinit Homo sapiens, felis catus ja Canis familiaris (Ensembl 96-julkaisusta) kartoitettiin täplähyeenan genomiin käyttäen tblastn (Blastall 2.2.26)27: ää parametreilla ”-e 1e-5”. Kohdistetut sekvenssit ja niiden kyselyproteiinit lähetettiin GeneWise – sovellukseen (versio 2.4.1) 28 tarkan liitoskohtauksen etsimistä varten. Lopullinen geenijoukko (22 747) kerättiin yhdistämällä ab initio-ja homologipohjaiset tulokset räätälöidyn putkilinjan avulla (taulukko 5).
Geenifunktioliitännät
Geenifunktiot määritettiin parhaan vastaavuuden mukaan, joka saatiin kohdistamalla käännetyt geenikoodaussekvenssit BLASTP: n avulla parametreilla ”-e 1e-5” SwissProt-ja TrEMBL-tietokantoihin (Uniprot release 2017-09). Geenien motiivit ja verkkotunnukset määritettiin InterProScan (v5)29: llä Proteiinitietokantoihin, joihin kuuluivat ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 ja PROSITE35. Kunkin geenin Ontologiatunnukset saatiin vastaavista SwissProt-ja TrEMBL-merkinnöistä. Kaikki geenit olivat linjassa KEGG-proteiineihin nähden, ja reitti, jossa geeni saattaa olla mukana, johdettiin Kegg-tietokannan vastaavista geeneistä36. Yhteenvetona voidaan todeta, että 22 166 (97,45%) ennustetuista proteiinia koodaavista geeneistä oli onnistuneesti merkitty vähintään yhteen kuudesta tietokannasta (Taulukko 6).
Geeniperheen Rakentaminen ja fylogenian rekonstruktio
saadaksemme tietoa Crocuta crocutan geeniperheiden fylogeneettisestä historiasta ja evoluutiosta ryhmitimme seitsemän lajin (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera Pardus, Panthera leo, Panthera tigris altaica) ja Homo sapiensin geeniperheiksi (Ensembl release-96, Panthera leo julkaisemattomista tiedoista) geeniperheiksi käyttäen Orthomcl (v2.0.9)37. Kahdeksan lajin proteiinia koodaavat geenit haettiin valitsemalla pisin transkriptio-isoformi kullekin geenille loppupään pairwise-tehtävää varten (graafin rakentaminen). Teimme BLASTP-haun kaikkien vertailulajien proteiinisekvensseistä E-arvon ollessa 1e-5. Geeniperheen rakentamisessa käytettiin MCL-algoritmia 38, jonka inflaatioparametri oli ”1.5”. Yhteensä 16 271 geeniperhettä C. crocuta, H. sapiens, F. catus, A. melanoleuca ryhmittyivät. Näillä neljällä lajilla oli 11 671 yhteistä geeniperhettä, kun taas 292 geeniperhettä, joissa oli 1 446 geeniä, olivat spesifisiä C. Crocutalle (Kuva. 5). Havaittavasti C. crocutan ja F. catus: n yhteiset geeniperimät olivat pienemmät kuin C. crocutan ja H. sapiensin yhteiset geeniperimät, mikä saattoi johtua siitä, että H. sapiensilla oli täydellisempi genomi ja merkintätapa.
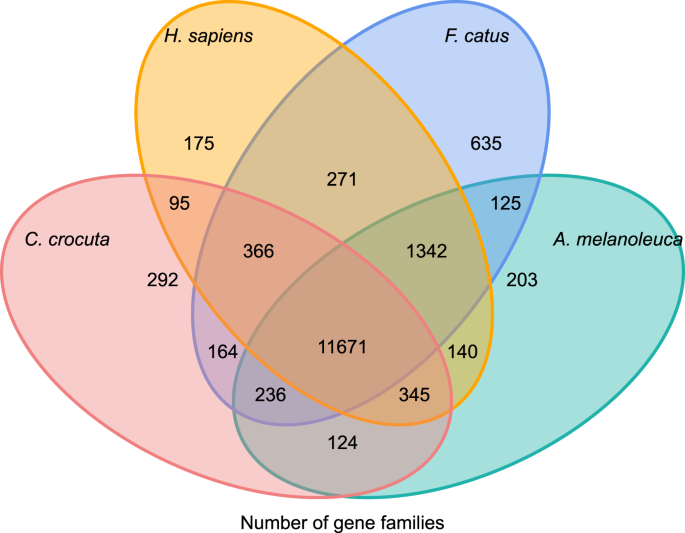
Venn-kaavio, jossa vertaillaan jaettuja ja ainutlaatuisia proteiinia koodaavia geenejä täplähyeenan, ihmisen, kotikissan ja kotimaisen koiran välillä ortologian analyysin perusteella.
tunnistimme 6 601 yksittäistä ortologista geeniä, joilla rekonstruoimme näiden kahdeksan lajin fylogeneettisen puun. Aminohapposekvenssien monisekvenssiset täsmäytykset kullekin geenille luotiin käyttäen lihasta (versio 3.8.31)39, ja karsittiin käyttäen Gblockeja (0.91 b)40, jolloin saavutettiin hyvin linjassa olevat alueet parametrien ”-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”kanssa. Suoritimme fylogeneettisen analyysin käyttäen maksimitodennäköisyysmenetelmää phyml: n (v3.0)41 mukaisesti käyttäen aminohapposubstituutiomallia JTT + G + I (Kuva. 6). Puun juuristo määritettiin minimoimalla koko puun korkeus Puupestin avulla (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Lopuksi, arvioimme ero aika joukossa kahdeksan lineages käyttäen MCMCTree alkaen PAML versio 4.4 software package42. Korvausasteen kalibrointiin käytettiin kahta fossiiliaineistoon perustuvaa prioriteettia, muun muassa Boreoeutheria (91-102 MYA) ja Carnivora (52-57 MYA)43. Aiempien tutkimusten mukaisesti täplähyeenaryhmät, joissa oli neljä lajia Felidae-heimosta kladissa, jossa määriteltiin alalahko Feliformia, joka erosi Caniformiasta (jota edustavat kotieläinkoira ja jättiläispanda) 53.9 Mya44.

C. crocutan fylogeneettinen puu ja seitsemän muuta lajia on rakennettu maksimitodennäköisyysmenetelmällä, joka perustuu 6 601: een kerta-ortologiaan. Divergenssiaika arvioitiin käyttämällä Aikapuun tietokannasta (http://www.timetree.org) johdettuja kahta kalibrointiprioria, joita merkitään punaromanssilla. Kaikki arvioidut eroajat on esitetty suluissa 95%: n luottamusvälillä.