脊髄性筋萎縮症
脊髄性筋萎縮症ファクトシートをダウンロード
MDAのCOVID-19応答について学ぶ
脊髄性筋萎縮症<3028><6291>脊髄性筋萎縮症(SMA)は、中枢神経系、末梢神経系、および随意筋運動(骨格筋)に影響を及ぼす遺伝性疾患です。
筋肉を制御する神経細胞の大部分は脊髄に位置しており、これは病気の名の中の脊髄という言葉を説明しています。 その主な効果は、これらの神経細胞からの信号を受信しない筋肉、上にあるので、SMAは筋肉です。 萎縮は、神経細胞によって刺激されていないときに筋肉に一般的に起こることである、小さくなるための医学用語です。
SMAは、脊髄の運動ニューロンと呼ばれる神経細胞の喪失を伴い、運動ニューロン疾患に分類されます。
SMAの最も一般的な形態(5番染色体SMA、またはSMN関連SMA)では、発症年齢、症状、および進行速度に広い変動性がある。 これらの違いを説明するために、しばしば常染色体劣性である染色体5関連SMAは、タイプ1から4に分類される。
SMAの症状が始まる年齢は、運動機能が影響を受ける程度とおおよそ相関しています。 出生時または乳児期に症状を示す子供は、典型的には最も低いレベルの機能を有する(タイプ1)。 重症度の低い経過を有する後発SMA(タイプ2および3、および十代または成人では、タイプ4)は、一般に、ますます高いレベルの運動機能と相関する。
詳細はSMAの形式を参照してください。
染色体5SMAは、”運動ニューロンの生存”のために、SMNと呼ばれる運動ニューロンタンパク質の欠乏によって引き起こされる。”このタンパク質は、その名前が示すように、正常な運動ニューロン機能に必要であると思われる。 SMNは運動ニューロンにおける遺伝子発現において極めて重要な役割を果たしている。 その欠乏は、SMN1と呼ばれる遺伝子の5番染色体上の遺伝的欠陥(突然変異)によって引き起こされる。 SMAと診断された患者内のSMN1遺伝子の最も一般的な突然変異は、エキソン7.1と呼ばれるセグメント全体の欠失であり、隣接するSMN2遺伝子は、これら二つの遺伝子の間に99%の同一性があるため、機能しないSMN1遺伝子を部分的に補うことができる。2
その他のまれな形態のSMA(非染色体5)は、SMN1.3以外の遺伝子の変異によって引き起こされます
まれな非染色体5リンクSMAの詳細な読み取りを含む詳
SMAの症状は何ですか?
SMAの症状は、軽度から重度の範囲の広いスペクトルをカバーしています。
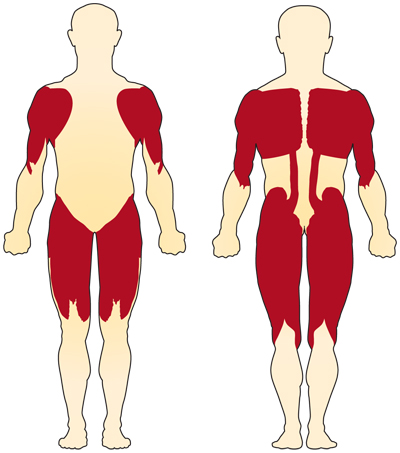
染色体5関連(SMN関連)SMAの主な症状は、随意筋の衰弱である。 最も影響を受ける筋肉は、肩、腰、太もも、背中上部など、身体の中心に最も近い筋肉です。 下肢は上肢よりも影響を受けているようで,深部けん反射は減少している。4
呼吸や嚥下に使用される筋肉に影響を与えると、特別な合併症が発生し、これらの機能に異常が生じます。 背中の筋肉が弱まると、脊髄湾曲が発達する可能性があります。
染色体5関連SMAで達成される発症年齢と運動機能のレベルには大きな変化があります。 これらは、運動ニューロンにどのくらいの機能的なSMNタンパク質が存在するかとほぼ相関しており、これは人が持っているSMN2遺伝子のコピーの数と相関している。 感覚的、精神的、および感情的機能は、染色体-5SMAでは完全に正常である。
SMAのいくつかの形態は、染色体5またはSMN欠乏症にリンクされていません。 これらの形態は、重症度および最も影響を受ける筋肉において大きく異なる。 ほとんどの形態が、染色体5関連の形態のような、大抵近位筋肉に影響を与える間、他の形態は大抵遠位筋肉に影響を与える存在します(ボディの中心
詳細は徴候と症状を参照してください。
SMAの進行は何ですか?
染色体5に関連するSMAでは、症状が後から始まり、SMNタンパク質が多いほど、疾患の経過は軽度になる可能性があります。 過去には、SMAを有する乳児は、典型的には2年以上生存しなかったが、今日、ほとんどの医師は現在、SMN関連SMAを連続体とみなし、発症年齢に厳密に基づいて平均寿命または衰弱について厳格な予測をしないことを好む。
SMAは乳児の死亡の最も一般的な遺伝的原因である。
SMAに関する研究の状況は何ですか?
研究は、この疾患の染色体5関連型に欠けているSMNタンパク質の体内産生を増加させるための戦略に焦点を当てている。 アプローチには、運動ニューロンが不利な状況で生き残るのを助ける方法が含まれます。
23、2016、米国食品医薬品局(FDA)は、SMAの治療のためのSpinraza(nusinersen)を承認しました。 Spinrazaは、SMAの根底にある欠陥を治療するように設計されているため、SMAの症状を減速、停止、または逆転させるのに有効である可能性があります。 詳細については、Spinrazaが承認されているを参照してください。
2019年5月、FDAは神経筋疾患の最初の遺伝子置換療法であるZolgensma(onasemnogene abeparvovac-xioi)を承認しました。 Zolgensmaは診断にpresymptomaticの人を含むSMN1遺伝子のbi対立遺伝子の突然変異のSMAとの2歳より若い小児科の患者の処置のための一度だけ静脈内の(静脈に)注入、で より多くの情報のために読まれたFDAは小児科の患者の背骨の筋萎縮の処置のためのAveXisのZolgensmaを承認する。
詳細は、研究、SMA:Full Speed AheadおよびIn Focus:Spinal Muscular Atrophy(SMA)を参照してください。 SMAと一緒に住んでいる家族の話については、強く、MDAのブログで私たちのSMAの物語を参照してください。
2020年8月、FDAは成人および生後2か月以上の小児のSMA治療のためのrisdiplam(ブランド名Evrysdi*)を承認しました。 Evysdiはsmn2″バックアップ”の遺伝子からの生産を高めることによってSMN蛋白質のレベルを増加するように設計されている口頭薬物です。
- 荻野S.&Wilson,R.B.脊髄性筋萎縮症(SMA)の遺伝子検査とリスク評価。 ヒト遺伝学(2002年)。 ドイ:10.1007/s00439-002-0828-x
- Lefebvre,S.et al. 脊髄性筋萎縮決定遺伝子の同定および特性評価。 セル(1995年)。 土井:10.1016/0092-8674(95)90460-3
- Darras,B.T.Non-5q脊髄性筋萎縮症:筋萎縮性側索硬化症(筋萎縮性側索硬化症)とは、筋萎縮性側索硬化症のことである。 神経学(2011年)。 土井:10.1212/WNL.0b013e3182267bd8
- Arnold,W.D.,Kassar,D.&Kissel,J.T.脊髄性筋萎縮症:新しい治療時代における診断と管理。 筋肉と神経(2015)。 土井:10.1002/mus.24497