Amyotrophie Spinale
Téléchargez notre Fiche d’information sur l’Amyotrophie spinale
En savoir plus sur la réponse de la MDA au COVID-19
Qu’est-ce que l’amyotrophie spinale?
L’amyotrophie spinale (SMA) est une maladie génétique affectant le système nerveux central, le système nerveux périphérique et les mouvements musculaires volontaires (muscle squelettique).
La plupart des cellules nerveuses qui contrôlent les muscles sont situées dans la moelle épinière, ce qui explique le mot colonne vertébrale dans le nom de la maladie. La SMA est musculaire car son effet principal est sur les muscles, qui ne reçoivent pas de signaux de ces cellules nerveuses. L’atrophie est le terme médical pour devenir plus petit, ce qui arrive généralement aux muscles lorsqu’ils ne sont pas stimulés par les cellules nerveuses.
La SMA implique la perte de cellules nerveuses appelées motoneurones dans la moelle épinière et est classée comme une maladie des motoneurones.
Dans la forme la plus courante de SMA (SMA du chromosome 5 ou SMA liée au SMN), il existe une grande variabilité de l’âge d’apparition, des symptômes et du taux de progression. Afin de tenir compte de ces différences, la SMA liée au chromosome 5, qui est souvent autosomique récessive, est classée dans les types 1 à 4.
L’âge auquel les symptômes de la SMA commencent est en corrélation avec le degré d’atteinte de la fonction motrice: Plus l’âge d’apparition est précoce, plus l’impact sur la fonction motrice est important. Les enfants qui présentent des symptômes à la naissance ou en bas âge ont généralement le niveau de fonctionnement le plus bas (type 1). SMA d’apparition tardive avec une évolution moins sévère (types 2 et 3, et chez les adolescents ou les adultes, type 4) est généralement en corrélation avec des niveaux de fonction motrice de plus en plus élevés.
Pour en savoir plus, voir Formes de SMA.
Quelles sont les causes de la SMA?
Le chromosome 5 SMA est causé par une carence en une protéine du motoneurone appelée SMN, pour « survie du motoneurone. »Cette protéine, comme son nom l’indique, semble être nécessaire au fonctionnement normal des motoneurones. Le SMN joue un rôle central dans l’expression des gènes dans les motoneurones. Sa déficience est causée par des défauts génétiques (mutations) sur le chromosome 5 d’un gène appelé SMN1. La mutation la plus courante dans le gène SMN1 chez les patients diagnostiqués avec SMA est une délétion d’un segment entier, appelé exon 7.1 Les gènes SMN2 voisins peuvent en partie compenser les gènes SMN1 non fonctionnels car il y a 99% d’identité entre ces deux gènes.2
D’autres formes rares de SMA (non-chromosome 5) sont causées par des mutations dans des gènes autres que SMN1.3
Pour plus d’informations, y compris une lecture détaillée sur les SMA rares non liées au chromosome 5, voir Formes de SMA et Causes /hérédité.
Quels sont les symptômes de la SMA?
Les symptômes de la SMA couvrent un large spectre, allant de légers à sévères.
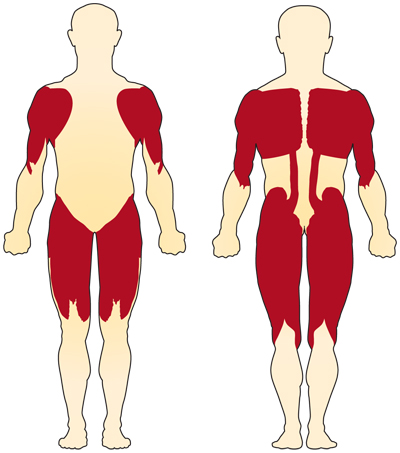
Le principal symptôme de la SMA liée au chromosome 5 (liée au SMN) est la faiblesse des muscles volontaires. Les muscles les plus touchés sont ceux les plus proches du centre du corps, tels que ceux des épaules, des hanches, des cuisses et du haut du dos. Les membres inférieurs semblent être affectés plus que les membres supérieurs et les réflexes tendineux profonds sont diminués.4
Des complications particulières surviennent si les muscles utilisés pour respirer et avaler sont affectés, entraînant des anomalies de ces fonctions. Si les muscles du dos s’affaiblissent, des courbures de la colonne vertébrale peuvent se développer.
Il y a beaucoup de variations dans l’âge d’apparition et le niveau de fonction motrice atteint dans la SMA liée au chromosome 5. Ceux-ci sont à peu près corrélés à la quantité de protéine SMN fonctionnelle présente dans les motoneurones, qui à son tour est en corrélation avec le nombre de copies de gènes SMN2 d’une personne. Le fonctionnement sensoriel, mental et émotionnel est tout à fait normal dans le chromosome 5 SMA.
Certaines formes de SMA ne sont pas liées à une déficience en chromosome 5 ou en SMN. Ces formes varient considérablement en gravité et dans les muscles les plus touchés. Alors que la plupart des formes, comme la forme liée au chromosome 5, affectent principalement les muscles proximaux, d’autres formes existent qui affectent principalement les muscles distaux (ceux qui sont plus éloignés du centre du corps) – du moins au début.
Pour en savoir plus, voir Signes et symptômes.
Quelle est la progression de la SMA?
Dans la SMA liée au chromosome 5, plus les symptômes commencent tard et plus il y a de protéines SMN, plus l’évolution de la maladie est susceptible d’être douce. Alors que dans le passé, les nourrissons atteints de SMA ne survivaient généralement pas plus de deux ans, aujourd’hui, la plupart des médecins considèrent désormais la SMA liée à la SMN comme un continuum et préfèrent ne pas faire de prédictions rigides sur l’espérance de vie ou la faiblesse basées uniquement sur l’âge d’apparition.
La SMA est la cause génétique la plus fréquente de mortalité chez les nourrissons.
Quel est l’état de la recherche sur la SMA?
La recherche s’est concentrée sur des stratégies visant à augmenter la production par l’organisme de la protéine SMN qui fait défaut dans les formes liées au chromosome 5 de la maladie. Les approches comprennent des méthodes pour aider les motoneurones à survivre dans des circonstances défavorables.
le déc. 23, 2016, la Food and Drug Administration (FDA) des États-Unis a approuvé Spinraza (nusinersen) pour le traitement de la SMA. Spinraza est conçu pour traiter le défaut sous-jacent de la SMA, ce qui signifie qu’il peut potentiellement être efficace pour ralentir, arrêter ou peut-être inverser les symptômes de la SMA. Pour en savoir plus, voir Spinraza est approuvé.
En mai 2019, la FDA a approuvé le Zolgensma (onasemnogene abeparvovac-xioi), la première thérapie de remplacement génique pour une maladie neuromusculaire. Zolgensma est une perfusion intraveineuse unique (dans la veine) destinée au traitement des patients pédiatriques de moins de 2 ans atteints de SMA présentant des mutations bi-alléliques du gène SMN1, y compris ceux qui sont présymptomatiques au diagnostic. Pour plus d’informations, lisez la FDA approuve le Zolgensma d’AveXis pour le traitement de l’Amyotrophie spinale chez les patients pédiatriques.
Pour en savoir plus, voir Recherche, SMA: Full Speed Ahead and In Focus: Amyotrophie spinale (SMA). Pour des histoires de familles vivant avec la SMA, consultez nos histoires de SMA sur Strongly, le blog de la MDA.
En août 2020, la FDA a approuvé risdiplam (nom de marque Evrysdi *) pour le traitement de la SMA chez les adultes et les enfants de deux mois ou plus. Evysdi est un médicament oral conçu pour augmenter les niveaux de la protéine SMN en améliorant la production à partir du gène « de secours » SMN2.
- Ogino, S. & Wilson, R. B. Genetic testing and risk assessment for spinal muscular amyotrophy (SMA). Génétique humaine (2002). doi: 10.1007/s00439-002-0828- x
- Lefebvre, S. et coll. Identification et caractérisation d’un gène déterminant l’amyotrophie spinale. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. Atrophies musculaires spinales non-5q: La soupe alphanumérique s’épaissit. Neurologie (2011). doi: 10.1212/WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. Amyotrophie spinale: Diagnostic et prise en charge dans une nouvelle ère thérapeutique. Muscle et nerf (2015). doi: 10.1002/ mus.24497