rdzeniowy zanik mięśni
Pobierz nasz rdzeniowy zanik mięśni arkusz informacyjny
dowiedz się więcej o odpowiedzi MDA na COVID-19
co to jest rdzeniowy zanik mięśni?
rdzeniowy zanik mięśni (SMA) jest chorobą genetyczną wpływającą na ośrodkowy układ nerwowy, Obwodowy układ nerwowy i dobrowolny ruch mięśni (mięśni szkieletowych).
większość komórek nerwowych, które kontrolują mięśnie, znajduje się w rdzeniu kręgowym, co stanowi słowo rdzeń w nazwie choroby. SMA jest muskularny, ponieważ jego główny wpływ jest na mięśnie, które nie odbierają sygnałów z tych komórek nerwowych. Atrofia jest terminem medycznym na coraz mniejsze, co zwykle dzieje się z mięśniami, gdy nie są stymulowane przez komórki nerwowe.
SMA polega na utracie komórek nerwowych zwanych neuronami ruchowymi w rdzeniu kręgowym i jest klasyfikowany jako choroba neuronu ruchowego.
w najczęstszej postaci SMA (chromosom 5 SMA lub SMN-related SMA), istnieje duża zmienność w wieku wystąpienia, objawów i szybkości progresji. W celu uwzględnienia tych różnic, SMA związane z chromosomem 5, który często jest autosomalny recesywny, klasyfikuje się do typów od 1 do 4.
wiek, w którym zaczynają się objawy SMA, z grubsza koreluje ze stopniem wpływu na funkcje motoryczne: im wcześniejszy wiek ich wystąpienia, tym większy wpływ na funkcje motoryczne. Dzieci, które wykazują objawy przy urodzeniu lub w okresie niemowlęcym, zwykle mają najniższy poziom funkcjonowania (typ 1). Późniejsza SMA o mniej ciężkim przebiegu (typy 2 i 3 oraz u nastolatków lub dorosłych Typ 4) na ogół koreluje z coraz wyższym poziomem funkcji motorycznych.
aby uzyskać więcej informacji, zobacz formy SMA.
co powoduje SMA?
chromosom 5 SMA jest spowodowany niedoborem białka neuronu ruchowego zwanego SMN, dla ” przetrwania neuronu ruchowego.”To białko, jak sama nazwa wskazuje, wydaje się być niezbędne do prawidłowego funkcjonowania neuronu ruchowego. SMN odgrywa kluczową rolę w ekspresji genów w neuronach ruchowych. Jego niedobór jest spowodowany wadami genetycznymi (mutacjami) na chromosomie 5 w genie zwanym SMN1. Najczęstszą mutacją w genie SMN1 u pacjentów z rozpoznaniem SMA jest delecja całego segmentu, zwana eksonem 7.1 sąsiednie geny SMN2 mogą częściowo zrekompensować niefunkcjonalne geny SMN1, ponieważ istnieje 99% tożsamości między tymi dwoma genami.2
inne rzadkie formy SMA (niezwiązane z chromosomem 5) są spowodowane mutacjami w genach innych niż SMN1.3
aby uzyskać więcej informacji, w tym szczegółowe czytanie na temat rzadkich, niezwiązanych z chromosomem 5 SMA, zobacz formy SMA i przyczyny/dziedziczenie.
jakie są objawy SMA?
objawy SMA obejmują szerokie spektrum, od łagodnego do ciężkiego.
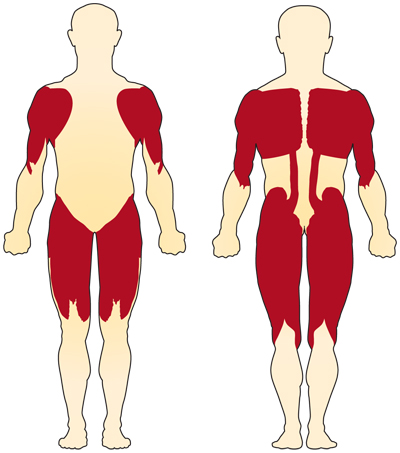
podstawowym objawem SMA związanego z chromosomem 5 (SMN-related) jest osłabienie mięśni dobrowolnych. Najbardziej dotknięte są mięśnie najbliższe centrum ciała, takie jak ramiona, biodra, uda i górna część pleców. Kończyny dolne wydają się być dotknięte bardziej niż kończyny górne, a Głębokie odruchy ścięgien są zmniejszone.
w przypadku zaburzeń czynności mięśni używanych do oddychania i połykania występują szczególne powikłania, co prowadzi do zaburzeń tych funkcji. Jeśli mięśnie pleców słabną, mogą rozwinąć się krzywizny kręgosłupa.
w SMA związanym z chromosomem 5 występuje duża zmienność wieku i poziomu funkcji motorycznych. Są one z grubsza skorelowane z ilością funkcjonalnego białka SMN obecnego w neuronach ruchowych, co z kolei koreluje z liczbą kopii genów SMN2. Funkcje sensoryczne, psychiczne i emocjonalne są całkowicie normalne w chromosomie-5 SMA.
niektóre formy SMA nie są związane z niedoborem chromosomu 5 lub SMN. Formy te różnią się znacznie w ciężkości i mięśni najbardziej dotkniętych. Podczas gdy większość form, takich jak forma związana z chromosomem 5, wpływa głównie na mięśnie bliższe, istnieją inne formy, które wpływają głównie na mięśnie dystalne (te dalej od centrum ciała)-przynajmniej na początku.
aby uzyskać więcej informacji, zobacz objawy podmiotowe i przedmiotowe.
jaka jest progresja SMA?
w SMA związanym z chromosomem 5, im później zaczynają się objawy i im więcej jest białka SMN,tym łagodniejszy jest przebieg choroby. Podczas gdy w przeszłości niemowlęta z SMA zazwyczaj nie przetrwały dłużej niż dwa lata, obecnie większość lekarzy uważa, że związane z SMN SMA jest kontinuum i woli nie przewidywać sztywnych przewidywań dotyczących średniej długości życia lub słabości w oparciu wyłącznie o wiek wystąpienia choroby.
SMA jest najczęstszą genetyczną przyczyną śmiertelności u niemowląt.
jaki jest status badań nad SMA?
badania koncentrują się na strategiach zwiększenia produkcji białka SMN w organizmie brakuje w chromosomie 5 związanych form choroby. Podejścia obejmują metody, które pomagają neuronom ruchowym przetrwać w niekorzystnych warunkach.
Food and Drug Administration (FDA) zatwierdziła Spinrazę (nusinersen) do leczenia SMA. Spinraza jest przeznaczony do leczenia podstawowej wady SMA, co oznacza, że potencjalnie może być skuteczny w spowolnieniu, zatrzymaniu lub odwróceniu objawów SMA. Aby uzyskać więcej informacji, zobacz Spinraza jest zatwierdzony.
w maju 2019 r.FDA zatwierdziła Zolgensma (onasemnogene abeparvovac-xioi), pierwszą genową terapię zastępczą w chorobie nerwowo-mięśniowej. Lek Zolgensma jest jednorazowym wlewem dożylnym (dożylnym) do leczenia pacjentów pediatrycznych w wieku poniżej 2 lat z SMA z mutacjami BI-allelicznymi w genie SMN1, w tym tych, którzy w momencie rozpoznania mają objawy poprzedzające rozpoznanie. Aby uzyskać więcej informacji, przeczytaj FDA zatwierdza Zolgensma AveXis w leczeniu rdzeniowego zaniku mięśni u pacjentów pediatrycznych.
aby uzyskać więcej informacji, zobacz badania, SMA: Full Speed Ahead and In Focus: Spinal Muscular Atrophy (SMA). Historie rodzin żyjących z SMA znajdziesz na blogu MDA.
w sierpniu 2020 r.FDA zatwierdziła risdiplam (Nazwa handlowa Evrysdi*) w leczeniu SMA u dorosłych i dzieci w wieku dwóch miesięcy lub starszych. Evysdi jest doustnym lekiem zaprojektowanym w celu zwiększenia poziomu białka SMN poprzez zwiększenie produkcji z „zapasowego” genu SMN2.
- Ogino, s. & Wilson, R. B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Human Genetics (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identyfikacja i charakterystyka rdzeniowego genu determinującego zanik mięśni. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. non-5Q spinal Muscular atrophies: the alfanumeric soup thickens. Neurologia (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. rdzeniowy zanik mięśni: Diagnostyka i postępowanie w nowej erze terapeutycznej. Muscle and Nerve (2015). doi: 10.1002 / mus.24497