Spinale Muskelatrophie
Laden Sie unser Fact Sheet zur spinalen Muskelatrophie herunter
Erfahren Sie mehr über die COVID-19-Reaktion von MDA
Was ist spinale Muskelatrophie?
Spinale Muskelatrophie (SMA) ist eine genetische Erkrankung, die das zentrale Nervensystem, das periphere Nervensystem und die freiwillige Muskelbewegung (Skelettmuskulatur) betrifft.
Die meisten Nervenzellen, die die Muskeln steuern, befinden sich im Rückenmark, was das Wort Wirbelsäule im Namen der Krankheit ausmacht. SMA ist muskulös, weil seine primäre Wirkung auf Muskeln ist, die keine Signale von diesen Nervenzellen empfangen. Atrophie ist der medizinische Begriff für das Verkleinern, was im Allgemeinen mit Muskeln passiert, wenn sie nicht von Nervenzellen stimuliert werden.
SMA beinhaltet den Verlust von Nervenzellen, die als Motoneuronen im Rückenmark bezeichnet werden, und wird als Motoneuronenerkrankung eingestuft.
Bei der häufigsten Form von SMA (Chromosom 5 SMA oder SMN-bezogene SMA) besteht eine große Variabilität in Bezug auf Erkrankungsalter, Symptome und Progressionsrate. Um diese Unterschiede zu berücksichtigen, wird die Chromosom 5-bezogene SMA, die häufig autosomal rezessiv ist, in die Typen 1 bis 4 eingeteilt.
Das Alter, in dem die SMA-Symptome beginnen, korreliert grob mit dem Grad, in dem die motorische Funktion betroffen ist: Je früher das Erkrankungsalter ist, desto größer ist der Einfluss auf die motorische Funktion. Kinder, die Symptome bei der Geburt oder im Säuglingsalter zeigen, haben typischerweise das niedrigste Funktionsniveau (Typ 1). Später einsetzende SMA mit einem weniger schweren Verlauf (Typen 2 und 3 und bei Teenagern oder Erwachsenen Typ 4) korreliert im Allgemeinen mit zunehmend höheren motorischen Funktionen.
Weitere Informationen finden Sie unter Formen von SMA.
Was verursacht SMA?
Chromosom 5 SMA wird durch einen Mangel an einem Motoneuronprotein namens SMN verursacht, für „Überleben von Motoneuron.“ Dieses Protein scheint, wie der Name schon sagt, für die normale Funktion von Motoneuronen notwendig zu sein. SMN spielt eine zentrale Rolle bei der Genexpression in Motoneuronen. Sein Mangel wird durch genetische Fehler (Mutationen) auf Chromosom 5 in einem Gen namens SMN1 verursacht. Die häufigste Mutation im SMN1-Gen bei Patienten, bei denen SMA diagnostiziert wurde, ist eine Deletion eines ganzen Segments, genannt Exon 7.1 Benachbarte SMN2-Gene können teilweise nicht funktionierende SMN1-Gene kompensieren, da zwischen diesen beiden Genen eine Identität von 99% besteht.2
Andere seltene Formen von SMA (nicht Chromosom 5) werden durch Mutationen in anderen Genen als SMN1 verursacht.3
Weitere Informationen, einschließlich detaillierter Informationen zu seltenen, nicht Chromosom 5-verknüpften SMA, finden Sie unter Formen von SMA und Ursachen / Vererbung.
Was sind die Symptome von SMA?
SMA-Symptome decken ein breites Spektrum ab, das von leicht bis schwer reicht.
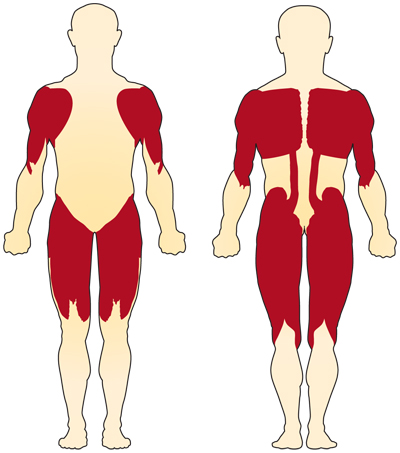
Das Hauptsymptom der Chromosom 5-bedingten (SMN-bedingten) SMA ist die Schwäche der freiwilligen Muskeln. Die am stärksten betroffenen Muskeln sind diejenigen, die der Körpermitte am nächsten liegen, z. B. Schultern, Hüften, Oberschenkel und oberer Rücken. Die unteren Gliedmaßen scheinen stärker betroffen zu sein als die oberen Gliedmaßen, und tiefe Sehnenreflexe sind vermindert.4
Besondere Komplikationen treten auf, wenn die zum Atmen und Schlucken verwendeten Muskeln betroffen sind, was zu Anomalien dieser Funktionen führt. Wenn die Rückenmuskulatur schwächer wird, können sich Krümmungen der Wirbelsäule entwickeln.
Bei Chromosom 5-bedingter SMA gibt es große Unterschiede im Erkrankungsalter und im Grad der motorischen Funktion. Diese korrelieren grob damit, wie viel funktionelles SMN-Protein in den Motoneuronen vorhanden ist, was wiederum damit korreliert, wie viele Kopien von SMN2-Genen eine Person hat. Sensorische, mentale und emotionale Funktionen sind bei Chromosom-5 SMA völlig normal.
Einige Formen von SMA sind nicht mit Chromosom 5 oder SMN-Mangel verbunden. Diese Formen variieren stark in der Schwere und in den am stärksten betroffenen Muskeln. Während die meisten Formen, wie die mit Chromosom 5 verwandte Form, hauptsächlich die proximalen Muskeln betreffen, gibt es andere Formen, die hauptsächlich die distalen Muskeln betreffen (die weiter vom Körperzentrum entfernt sind) — zumindest am Anfang.
Weitere Informationen finden Sie unter Anzeichen und Symptome.
Was ist das Fortschreiten von SMA?
Bei Chromosom 5-bedingter SMA ist der Krankheitsverlauf wahrscheinlich umso milder, je später die Symptome beginnen und je mehr SMN-Protein vorhanden ist. Während in der Vergangenheit Säuglinge mit SMA in der Regel nicht mehr als zwei Jahre überlebten, betrachten die meisten Ärzte SMN-bedingte SMA heute als Kontinuum und ziehen es vor, keine starren Vorhersagen über die Lebenserwartung oder Schwäche zu treffen, die ausschließlich auf dem Erkrankungsalter basieren.
SMA ist die häufigste genetische Todesursache bei Säuglingen.
Wie ist der Stand der SMA-Forschung?
Die Forschung hat sich auf Strategien konzentriert, um die körpereigene Produktion des SMN-Proteins zu erhöhen, das in den Chromosom-5-verwandten Formen der Krankheit fehlt. Ansätze umfassen Methoden, um Motoneuronen zu helfen, unter widrigen Umständen zu überleben.
Am Dez. 23, 2016, die US Food and Drug Administration (FDA) genehmigt Spinraza (nusinersen) für die Behandlung von SMA. Spinraza wurde entwickelt, um den zugrunde liegenden Defekt bei SMA zu behandeln, was bedeutet, dass es möglicherweise wirksam sein kann, um die Symptome von SMA zu verlangsamen, zu stoppen oder vielleicht umzukehren. Weitere Informationen finden Sie unter Spinraza ist zugelassen.
Im Mai 2019 genehmigte die FDA Zolgensma (Onasemnogen abeparvovac-xioi), die erste Genersatztherapie für eine neuromuskuläre Erkrankung. Zolgensma ist eine einmalige intravenöse (in die Vene) Infusion zur Behandlung von pädiatrischen Patienten unter 2 Jahren mit SMA mit biallelischen Mutationen im SMN1-Gen, einschließlich solcher, die bei der Diagnose präsymptomatisch sind. FDA genehmigt Zolgensma von AveXis zur Behandlung von spinaler Muskelatrophie bei pädiatrischen Patienten.
Weitere Informationen finden Sie unter Research, SMA: Full Speed Ahead and In Focus: Spinal Muscular Atrophy (SMA). Geschichten von Familien, die mit SMA leben, finden Sie in unseren SMA-Geschichten auf
Im August 2020 genehmigte die FDA Risdiplam (Markenname Evrysdi *) zur Behandlung von SMA bei Erwachsenen und Kindern ab zwei Monaten. Evysdi ist ein orales Medikament, das entwickelt wurde, um den Spiegel des SMN-Proteins zu erhöhen, indem die Produktion aus dem „Backup“ -Gen SMN2 gesteigert wird.
- Ogino, S. & Wilson, R. B. Gentests und Risikobewertung für spinale Muskelatrophie (SMA). Humangenetik (2002). ust-idnr.:10.1007/s00439-002-0828- x
- Lefebvre, S. et al. Identifizierung und Charakterisierung eines spinalen Muskelatrophie-bestimmenden Gens. Zelle (1995). doi:10.1016/0092-8674(95)90460-3
- Darras, B. T. Non-5q spinale Muskelatrophien: Die alphanumerische Suppe verdickt sich. Neurologie (2011). Ursprungsbezeichnung:10.1212/WNL.0b013e3182267bd8
- Arnold, WD, Kassar, D. & Kissel, JT Spinale Muskelatrophie: Diagnose und Behandlung in einer neuen therapeutischen Ära. Muskel und Nerven (2015). doi:10.1002/mus.24497