Spinal muskelatrofi
Hent vores Spinal muskelatrofi faktaark
Lær om MDAS COVID-19-respons
hvad er spinal muskelatrofi?
Spinal muskelatrofi (SMA) er en genetisk sygdom, der påvirker centralnervesystemet, det perifere nervesystem og frivillig muskelbevægelse (skeletmuskulatur).
de fleste af nervecellerne, der styrer musklerne, er placeret i rygmarven, som tegner sig for ordet spinal i sygdommens navn. SMA er muskuløs, fordi dens primære virkning er på muskler, som ikke modtager signaler fra disse nerveceller. Atrofi er den medicinske betegnelse for at blive mindre, hvilket er hvad der generelt sker med muskler, når de ikke stimuleres af nerveceller.
SMA involverer tab af nerveceller kaldet motorneuroner i rygmarven og klassificeres som en motorneuronsygdom.
i den mest almindelige form for SMA (kromosom 5 SMA eller SMN-relateret SMA) er der stor variation i begyndelsesalder, symptomer og progressionshastighed. For at tage højde for disse forskelle klassificeres kromosom 5-relateret SMA, som ofte er autosomal recessiv, i type 1 til 4.
den alder, hvor SMA-symptomer begynder, korrelerer groft med, i hvilken grad motorfunktionen påvirkes: jo tidligere begyndelsesalderen er, desto større er virkningen på motorfunktionen. Børn, der viser symptomer ved fødslen eller i spædbarnet, har typisk det laveste funktionsniveau (type 1). Senere debut SMA med et mindre alvorligt forløb (type 2 og 3 og hos teenagere eller voksne, type 4) korrelerer generelt med stadig højere niveauer af motorisk funktion.
For mere, Se former for SMA.
Hvad forårsager SMA?
kromosom 5 SMA er forårsaget af en mangel på et motorneuronprotein kaldet SMN, til “overlevelse af motorneuron.”Dette protein, som navnet antyder, synes at være nødvendigt for normal motorisk neuronfunktion. SMN spiller en central rolle i genekspression i motoriske neuroner. Dens mangel er forårsaget af genetiske fejl (mutationer) på kromosom 5 i et gen kaldet SMN1. Den mest almindelige mutation i SMN1-genet hos patienter diagnosticeret med SMA er en deletion af et helt segment, kaldet ekson 7.1 nærliggende SMN2-gener kan delvis kompensere for ikke-funktionelle SMN1-gener, da der er 99% identitet mellem disse to gener.2
andre sjældne former for SMA (ikke-kromosom 5) er forårsaget af mutationer i andre gener end SMN1.3
For mere information, herunder detaljeret læsning af sjældne, ikke-kromosom 5-forbundne SMA, se former for SMA og årsager/arv.
hvad er symptomerne på SMA?
SMA-symptomer dækker et bredt spektrum, der spænder fra mild til svær.
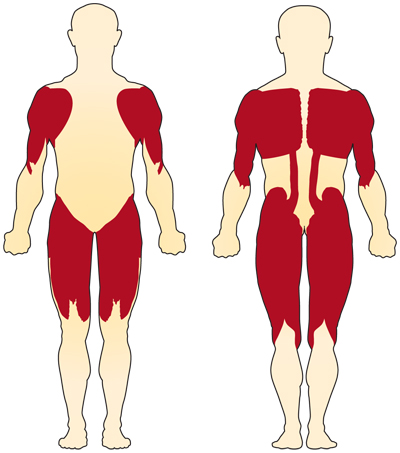
det primære symptom på kromosom 5-relateret (SMN-relateret) SMA er svaghed i de frivillige muskler. De mest berørte muskler er dem, der er tættest på midten af kroppen, såsom skuldre, hofter, lår og øvre ryg. De nedre lemmer ser ud til at være påvirket mere end de øvre lemmer, og dybe senereflekser reduceres.4
særlige komplikationer opstår, hvis musklerne, der bruges til vejrtrækning og indtagelse, påvirkes, hvilket resulterer i abnormiteter i disse funktioner. Hvis musklerne i ryggen svækkes, kan spinalkurvaturer udvikle sig.
der er stor variation i begyndelsesalderen og niveauet af motorisk funktion opnået i kromosom 5-relateret SMA. Disse er groft korreleret med, hvor meget funktionelt SMN-protein der er til stede i motorneuronerne, hvilket igen korrelerer med hvor mange kopier af SMN2-gener en person har. Sensorisk, mental og følelsesmæssig funktion er helt normal i kromosom – 5 SMA.
nogle former for SMA er ikke knyttet til kromosom 5 eller SMN-mangel. Disse former varierer meget i sværhedsgrad og i de mest berørte muskler. Mens de fleste former, som den kromosom 5-relaterede form, hovedsageligt påvirker de proksimale muskler, findes der andre former, der hovedsageligt påvirker de distale muskler (dem længere væk fra kroppens centrum) — i det mindste i begyndelsen.
For mere, se tegn og symptomer.
Hvad er udviklingen af SMA?
i kromosom 5-relateret SMA, jo senere symptomerne begynder, og jo mere SMN-protein der er, jo mildere er sygdomsforløbet sandsynligvis. Mens i fortiden, spædbørn med SMA typisk ikke overlevede mere end to år, i dag betragter de fleste læger nu SMN-relateret SMA som et kontinuum og foretrækker ikke at komme med stive forudsigelser om forventet levealder eller svaghed baseret strengt på begyndelsesalderen.
SMA er den mest almindelige genetiske årsag til dødelighed hos spædbørn.
hvad er status for forskning på SMA?
forskning har fokuseret på strategier til at øge kroppens produktion af SMN-proteinet, der mangler i de kromosom 5-relaterede former for sygdommen. Tilgange inkluderer metoder til at hjælpe motoriske neuroner med at overleve under ugunstige omstændigheder.
Den Dec. 23, 2016, den amerikanske Food and Drug Administration (FDA) godkendt Spinrasa (nusinersen) til behandling af SMA. Det er designet til at behandle den underliggende defekt i SMA, hvilket betyder, at det potentielt kan være effektivt til at bremse, stoppe eller måske vende symptomerne på SMA. For mere, se Spinra er godkendt.
i maj 2019 godkendte FDA den første generstatningsterapi for en neuromuskulær sygdom. Tsolgensma er en engangs intravenøs (i venen) infusion til behandling af pædiatriske patienter yngre end 2 år med SMA med bi-alleliske mutationer i SMN1-genet, herunder dem, der er presymptomatiske ved diagnose. For mere information læs FDA godkender Aveksis til behandling af Spinal muskelatrofi hos pædiatriske patienter.
For mere, se Forskning, SMA: fuld fart fremad og i fokus: Spinal muskelatrofi (SMA). For historier om familier, der bor hos SMA, se vores SMA-historier på stærkt, MDA-bloggen.
i August 2020 godkendte FDA risdiplam (mærkenavn Evrysdi*) til behandling af SMA hos voksne og børn, der er to måneder eller ældre. Evysdi er en oral medicin designet til at øge niveauerne af SMN-proteinet ved at øge produktionen fra SMN2 “backup” – genet.
- Ogino, S. & R. B. genetisk testning og risikovurdering for spinal muskelatrofi (SMA). Human Genetik (2002). doi: 10.1007 / s00439-002-0828-
- Lefebvre, S. et al. Identifikation og karakterisering af et spinal muskelatrofi-bestemmende gen. Celle (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. ikke-5K spinal muskulære atrofier: den alfanumeriske suppe tykner. Neurologi (2011). doi: 10.1212 / VNL.0b013e3182267bd8
- Arnold, V. D., Kassar, D. & Kissel, J. T. Spinal muskelatrofi: diagnose og styring i en ny terapeutisk æra. Muskel og Nerve (2015). doi: 10.1002 / mus.24497