- Atrofia Muscular Espinhal
- o Que é a atrofia muscular espinhal?A atrofia muscular espinhal (SMA) é uma doença genética que afecta o sistema nervoso central, o sistema nervoso periférico e o movimento muscular voluntário (músculo esquelético).
- o que causa SMA?
- quais são os sintomas do SMA?
- Qual é a progressão de SMA?
- Qual é o estado da investigação sobre SMA?
Atrofia Muscular Espinhal
Baixe o nosso Atrofia Muscular Espinhal Fato de Folha
Aprender sobre MDA do COVID-19 de resposta
o Que é a atrofia muscular espinhal?A atrofia muscular espinhal (SMA) é uma doença genética que afecta o sistema nervoso central, o sistema nervoso periférico e o movimento muscular voluntário (músculo esquelético).
A maior parte das células nervosas que controlam os músculos estão localizadas na medula espinhal, o que explica a palavra spinal em nome da doença. O SMA é muscular porque o seu efeito primário é nos músculos, que não recebem sinais destas células nervosas. Atrofia é o termo médico para ficar menor, que é o que geralmente acontece com os músculos quando eles não são estimulados pelas células nervosas.
SMA envolve a perda de células nervosas chamadas neurônios motores na medula espinhal e é classificado como uma doença neuronal motor.
na forma mais comum de SMA (cromossoma 5 SMA, ou SMN relacionado SMA), existe uma grande variabilidade na idade de início, sintomas e taxa de progressão. A fim de ter em conta estas diferenças, o SMA relacionado com o cromossoma 5, que muitas vezes é autossômico recessivo, é classificado nos tipos 1 a 4.
a idade em que os sintomas de SMA começam correlaciona-se aproximadamente com o grau em que a função motora é afetada: quanto mais cedo a idade de início, maior o impacto na função motora. As crianças que apresentam sintomas no nascimento ou na infância normalmente têm o nível mais baixo de funcionamento (Tipo 1). Um SMA de início posterior com um curso menos grave (tipos 2 e 3, e em adolescentes ou adultos, Tipo 4) geralmente correlaciona-se com níveis cada vez mais elevados de função motora.
para mais, ver formas de SMA.
o que causa SMA?
o Cromossoma 5 SMA é causado por uma deficiência de uma proteína de neurônio motor chamada SMN, para a “sobrevivência do neurônio motor”.”Esta proteína, como seu nome indica, parece ser necessária para a função normal do neurônio motor. A SMN desempenha um papel fundamental na expressão genética nos neurônios motores. Sua deficiência é causada por falhas genéticas (mutações) no cromossomo 5 em um gene chamado SMN1. A mutação mais comum no gene SMN1 em pacientes diagnosticados com SMA é uma exclusão de um segmento inteiro, chamado exon 7.1 genes vizinhos SMN2 pode em parte compensar genes SMN1 não-funcionais, uma vez que há 99% de identidade entre estes dois genes.2
Outras formas raras de SMA (não cromossoma 5) são causadas por mutações em genes de outros que SMN1.3
Para mais informações, incluindo leitura detalhada sobre raro, não cromossoma 5-vinculado SMA, consulte Formas de SMA e Faz com que/Herança.
quais são os sintomas do SMA?
os sintomas SMA abrangem um amplo espectro, que vão de ligeira a grave.
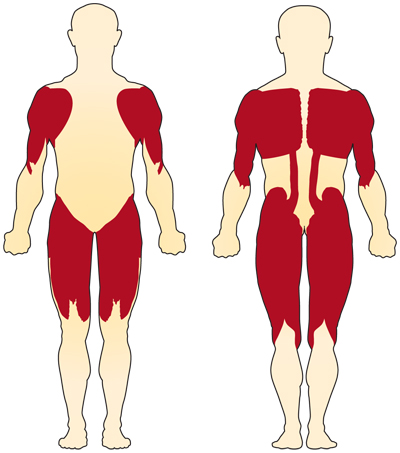
o principal sintoma do SMA relacionado com o cromossoma 5 (relacionado com o SMN) é a fraqueza dos músculos voluntários. Os músculos mais afetados são os mais próximos ao centro do corpo, como os dos ombros, quadris, coxas e parte superior das costas. Os membros inferiores parecem ser mais afectados do que os membros superiores e os reflexos profundos dos tendões diminuem.4
ocorrem complicações especiais se os músculos utilizados para respirar e engolir forem afectados, resultando em anomalias nestas funções. Se os músculos das costas enfraquecerem, as curvaturas espinais podem desenvolver-se.
há uma grande variação na idade de início e nível de função motora atingido no cromossoma 5-relacionado SMA. Estas estão aproximadamente correlacionadas com a quantidade funcional de proteína SMN presente nos neurônios motores, que por sua vez se correlaciona com quantas cópias de genes SMN2 uma pessoa tem. O funcionamento sensorial, mental e emocional é completamente normal no cromossoma 5 SMA.
algumas formas de SMA não estão relacionadas com a deficiência do cromossoma 5 ou SMN. Estas formas variam muito em gravidade e nos músculos mais afetados. Enquanto a maioria das formas, como a forma 5-relacionada ao cromossomo, afetam principalmente os músculos proximais, outras formas existem que afetam principalmente os músculos distais (aqueles mais distantes do centro do corpo) — pelo menos no início.
para mais, ver sinais e sintomas.
Qual é a progressão de SMA?
no cromossoma 5-relacionado SMA, quanto mais tarde os sintomas começam e quanto mais proteína SMN existe, mais suave é o curso da doença. Enquanto no passado, os bebês com SMA tipicamente não sobreviviam mais de dois anos, hoje a maioria dos médicos consideram o SMA relacionado com SMN como um continuum e preferem não fazer previsões rígidas sobre a expectativa de vida ou fraqueza baseada estritamente na idade de início.
SMA é a causa genética mais comum de mortalidade em lactentes.
Qual é o estado da investigação sobre SMA?
a investigação centrou-se em estratégias para aumentar a produção do organismo da proteína SMN que não possui as formas do cromossoma 5 relacionadas com a doença. Abordagens incluem métodos para ajudar os neurônios motores a sobreviver em circunstâncias adversas.
Em Dez. 23, 2016, A U. S. Food and Drug Administration (FDA) aprovou Spinraza (nusinersen) para o tratamento da SMA. Spinraza é projetado para tratar o defeito subjacente na SMA, o que significa que pode ser potencialmente eficaz na desaceleração, parada, ou talvez reverter os sintomas da SMA. Para mais, veja Spinraza é aprovado.
em maio de 2019, a FDA aprovou Zolgensma (onasemnogene abeparvovac-xioi), a primeira terapia de substituição de genes para uma doença neuromuscular. O Zolgensma é uma perfusão intravenosa única (na veia) para o tratamento de doentes pediátricos com menos de 2 anos de idade com SMA com mutações bi-alélicas no gene SMN1, incluindo aqueles que são pré-assintomáticos no diagnóstico. Para mais informações leia a FDA aprova o Zolgensma do AveXis para o tratamento da atrofia Muscular espinhal em doentes pediátricos.
para mais, ver pesquisa, SMA: velocidade máxima à frente e em Foco: atrofia Muscular espinhal (SMA). Para histórias de famílias que vivem com SMA, veja nossas histórias Sma em Strongly, o blog MDA.
em agosto de 2020, a FDA aprovou o risdiplam (nome de marca Evrysdi*) para o tratamento de SMA em adultos e crianças com dois ou mais meses de idade. Evysdi é um medicamento oral projetado para aumentar os níveis da proteína SMN, aumentando a produção do gene SMN2 “backup”.
- Ogino, S. & Wilson, R. B. ensaios genéticos e avaliação dos riscos de atrofia muscular espinhal (SMA). Human Genetics (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identificação e caracterização de um gene que determina a atrofia muscular espinhal. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. Não-5q atrofias musculares espinhais: a sopa alfanumérica engrossa. Neurology (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. atrofia muscular da coluna vertebral: diagnóstico e gestão numa nova era terapêutica. Muscle and Nerve (2015). doi: 10.1002 / mus.24497