Sample collection,library construction and sequencing
のゲノムDNAを、C.crocutaの雄標本から得た(NCBI taxonomy ID:9678;Fig. 1)San Diego Zoo Institute for Converation Research,USAのFrozing Zoo(登録商標)に保存した(Frozing Zoo ID:KB4 5 2 6)。
ゲノムDNAをフェノール-クロロホルムを用いて抽出し、エタノール沈殿13を用いて精製した。 抽出されたDNAを実行し、1上で可視化した。高分子量DNAの存在があるように確認するために1x TBEの緩衝の5%agaroseのゲルの操業。 0蛍光光度計(Thermo Fisher Scientific、USA)上でDNA濃度および純度を定量した後、Bgi−Shenken、中国に出荷した。 我々は、372μ gのゲノムDNAの合計を得た0.418μ g/μ l Nanodrop2000と0.245–0.399μ g/μ l量子ビット2.0蛍光光度計を使用して四つの複製測定値に基づいて使用してμ g/μ lの濃度 純度の260/280比は1.95であった。 次に,シトクロムb(Cytb)遺伝子を用いて試料をバーコード化した。 次に、勾配ライブラリ戦略に従って、170bp、500bp、800bp、2kbp、5kbp、10kbp、20kbpの挿入サイズの長さを持つ13の挿入サイズライブラリを構築しました。 私たちはHiSeqを使用しました。 2000sequencer(Illumina,USA)To sequence Paired-End(PE)reads for each library across14lane. 合計で約299Gbの生データが13のライブラリから生成され、149.25のシーケンス深度(カバレッジ)を達成しました(表1)。
品質管理
誤組立エラーを最小限に抑えるために、de novo genome assemblyの前に生の読み取りを以下の二つの基準に従ってフィルタリングしました。 最初に、アダプタシーケンスに整列した10bpを超える読み取り(<=3bpの不一致を可能にする)が削除されました。 第二に、1 0以下の品質値を有する塩基の4 0%を有するリードを廃棄した。 最後に、190.4Gのデータを95.2のカバレッジで得ました(表2)。
ゲノムサイズの推定
三つの短い挿入ライブラリ(170bpの二つと500bpの一つ)は、k-mer解析によってゲノムサイズとゲノム全体のヘテロ接合性を推定するため 合計約385MのPE読み取りは、k-mer周波数を計算するためにjellyfish14に提出されました。 次に、パラメータ”k=17;length=100;max coverage=1000″を用いて、K-mer分布をGenomescope7によって例示した。 我々は、推定ゲノムサイズ2,003,681,234bp、および0.325%のヘテロ接合性を得た(図。 2).

17-merのゲノムサイズの推定値。 X軸は深さ(X)であり、y軸は、その深さにおける周波数を全てのカバレッジ深さの合計周波数で割った割合である。 配列誤り率、ヘテロ接合率、およびゲノムの繰り返し率を考慮せずに、17mer分布はポアソン分布に近似する必要があります。
ゲノムの組み立てと評価
SOAPdenovo(V1.短い挿入サイズデータのフィルタリングおよび大きな挿入サイズデータの小さなピークを除去した後に、ゲノムde novoを組み立てるために、06)15を用いた。 SOAPdenovoアセンブリアルゴリズムには、三つの主要なステップが含まれていました。 (1)コンティグ構造:短い挿入サイズのライブラリデータをk-mersに分割し、チップを除去し、気泡をマージし、接続の低カバレッジを除去し、小さな繰り返しを除去することによって簡素化されたde Bruijnグラフを用いて構築した。 我々は、コンティグN50 2,104bp、および全長2,295,545,898bpで得られ、k-merパスを接続することによってコンティグ配列を得た。 (2)足場の構造:私達は連続したすべての使用可能な読まれる再調整によってすべての一直線に並べられた対端の読まれるの80%を得ました。 次に、連続の各ペア間の共有対端関係の量を計算し、一貫性と競合する対端の割合を重み付けし、足場を段階的に構築しました。 その結果、我々はn50 7,168,038bpと足場を得た、と全長2,355,303,269bp短い挿入サイズのペアエンドから、長い遠いペアエンドに。 (3)ギャップ閉鎖:構築された足場内のギャップを埋めるために、我々はこれらの収集された読み取りのために再びローカルアセンブリを行うために読 要約すると、我々は、足場内のギャップの87.7%、または合計ギャップの長さの85.8%を閉じました。 Contig N50サイズは2,104bpから21,301bpに増加しました(表3)。 足場アセンブリサイズは2,355,303,269bpであり、これはストライプhyaena、Hyaena hyaena11(NCBI accession:Asm300989V1)について報告された2,374,716,107bpのアセンブリベースのゲノムサイズに近い。 我々はまた、この種の配列決定された最初のミトコンドリアゲノムと同様に、16,858bpの長さを有するMitoZプログラム16を用いて、斑点を付けられたハイエナのミト
ドラフトゲノムの評価は、BUSCO(バージョン3.1.0)17を使用してシングルコピー orthologの完全性を見て、4,104シングルコピー orthologグループを含むMammaliaodb9データベースに対して検索することによって行われた。 オルソログの95.5%の合計は、斑点ハイエナゲノムアセンブリの全体的な高品質を示す、完全な、断片化として2.5%と欠落として2.0%として同定されました。 短い足場(<1k)の99.95%が1しか保有していないことを考えると。総ゲノム長の2%は、我々は反復要素と遺伝子機能注釈を含む下流の分析のためにこれらの足場を除外しました。
反復要素注釈
タンデムリピートとtransposable要素(TE)の両方を検索し、C.crocutaゲノム全体で同定しました。 タンデムリピートは、タンデムリピートファインダー(TRF、v4.07)18を使用して同定され、transposable要素(TEs)は、相同性ベースとde novoアプローチの組み合わせによって同定された。 相同性ベースの予測のために、我々はRepeatMaskerバージョン4.0を使用しました。0 0 0 1」を設定してRepeatproteinmask(Repeatmaskerパッケージ内のプログラム)を設定して、既知の反復に基づいてヌクレオチドおよびアミノ酸レベルでTesを検索する(図6 1 9)。 3). RepeatMaskerは、Repbase21.10dataset20を組み合わせたカスタムライブラリを使用してDNAレベルの同定に適用されました。 タンパク質レベルでは、RepeatProteinMaskは、TEタンパク質データベースに対してRMBlastを実行するために使用されました。 Ab initio予測の場合、RepeatModeler(v1.0.8)21およびLTR_FINDING(v1.06)22をde novo repeat libraryを構築するために適用した。 ライブラリ内の汚染およびマルチコピー配列を除去し、残りの配列をSwissProtデータベースへのアライメント後のBLAST結果に従って分類した。 このライブラリーに基づいて、本発明者らは、相同T EをマスクするためにRepeatmaskerを使用し、それらを分類した(図1 0A)。 4). 全体として、全ゲノムの35.29%を含む、斑点状のハイエナで合計826Mbの反復要素が同定された(表4)。

相同性に基づく予測に基づくクロクタクロクタゲノムアセンブリにおけるトランスポーザブルエレメント(TE)の各タイプの発散率の分布。 相同性ベースの方法を用いてゲノム中の同定されたTEsとRepbase database20のコンセンサス配列との間の発散率を計算した。

ab initio予測に基づくCrocuta crocutaゲノムアセンブリにおけるTEの各タイプの発散率の分布。 Ab initio予測によってゲノム中の同定されたTEsと予測されたTEライブラリ中のコンセンサス配列との間の発散率を計算した(方法を参照)。
タンパク質コード遺伝子注釈
我々は、タンパク質コード遺伝子だけでなく、スプライシングサイトと代替スプライシングアイソフォームを注釈するためにab initio予測とホモログベースのアプローチを使用しました。 Ab initio予測は、AUGUSTUS(バージョン2.5.5)23、GENSCAN24、GlimmerHMM(バージョン3.0.4)25、およびSNAP(バージョン2006-07-28)26をそれぞれ使用して、ヒト、家畜犬、および家畜猫からの遺伝子モデルを使 この方法によって合計22,789個の遺伝子が同定された。 Homo sapiens、Felis catusおよびCanis familiaris(Ensembl96リリースから)の相同タンパク質は、パラメータ”-e1e-5″を持つtblastn(Blastall2.2.26)27を使用して斑点を付けられたハイエナゲノムにマッピングされた。 整列された配列だけでなく、それらのクエリタンパク質は、正確なスプライスアライメントを検索するためにGeneWise(バージョン2.4.1)28に提出されました。 最終的な遺伝子セット(22,747)は、カスタマイズされたパイプラインを使用してab initioおよびホモログベースの結果をマージすることによって収集された(表5)。
遺伝子機能注釈
遺伝子機能は、BLASTPを用いて翻訳された遺伝子コード配列をパラメータ”-e1e-5″で整列させた最良の一致に従って、SwissProtおよびTrEMBLデータベース(Uniprot release2017-09)に割り当てられた。 遺伝子のモチーフとドメインは、Prodom30、PRINTS31、Pfam32、SMART33、PANTHER34とPROSITE35を含むタンパク質データベースに対してInterProScan(v5)29によって決定されました。 各遺伝子の遺伝子オントロジー Idは、対応するSwissProtおよびTrEMBLエントリから得られた。 すべての遺伝子はKEGGタンパク質に対して整列し、遺伝子が関与する可能性のある経路は、KEGGデータベース36の一致した遺伝子から誘導された。 要約すると、予測されたタンパク質コード遺伝子の22,166(97.45%)は、六つのデータベースの少なくとも一つによって首尾よく注釈された(表6)。
遺伝子ファミリーの構築と系統発生の再構成
Crocuta crocutaの遺伝子ファミリーの系統発生の歴史と進化への洞察を得るために、我々は七種(Felis catus、Canis familiaris、Ailuropoda melanoleuca、Crocuta crocuta、Panthera pardus、Panthera leo、Panthera tigris altaica)とHomo sapiensの遺伝子配列をアウトグループとしてクラスター化した(Ensembl release-96、未発表データからPanthera leo)。Orthomcl(v2.0.9)37. 下流のペアワイズ割り当て(グラフ構築)のために各遺伝子の最長転写アイソフォームを選択することにより、八種のタンパク質コード遺伝子を検索した。 我々は、1e-5のE値カットオフで、すべての参照種のタンパク質配列上のすべてに対してすべてのBLASTP検索を行った。 遺伝子ファミリーの構築は、インフレーションパラメータが’1.5’のMCLアルゴリズムを採用した。 C.crocuta、H.sapiens、F.catus、A.melanoleucaの16,271遺伝子ファミリーの合計がクラスタ化されました。 これらの4種で共有されている遺伝子ファミリーは11,671個であり、1,446個の遺伝子を含む292個の遺伝子ファミリーはC.Crocutaに特異的であった(図1)。 5). 明らかに、c.crocutaとF.catusが共有した遺伝子ファミリーは、C.crocutaとH.sapiensが共有した遺伝子ファミリーよりも小さく、H.sapiensがより完全なゲノムと注釈を持っていたことに起因する可能性がある。
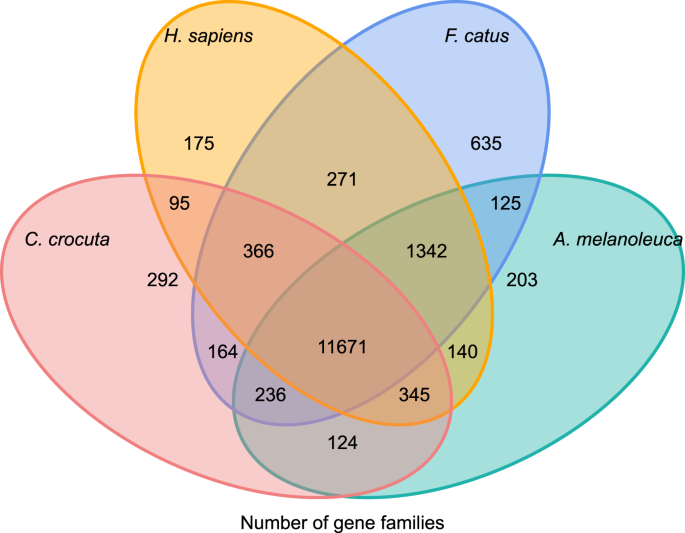
オーソロジー解析に基づいて、斑点ハイエナ、ヒト、国内の猫と国内の犬の間で共有され、ユニークなタンパク質コード遺伝子の比較を示すベン図。
我々は、八種の系統樹を再構築するために6,601シングルコピー orthologous遺伝子を同定しました。 各遺伝子のアミノ酸配列の複数の配列アライメントは、筋肉(バージョン3.8.31)39を使用して生成され、Gblocks(0.91b)40を使用してトリミングされ、パラメータ”-t=p-b3=8-b4=10-b5=n-e=-st”を有するよく整列された領域を達成した。 本発明者らは、アミノ酸置換のためのJTT+G+Iモデルを用いて、PhyML(v3.0)41に実装されている最尤法を用いて系統発生解析を行った(図3)。 6). ツリーのルートは、Treebest(v1)を介してツリー全体の高さを最小化することによって決定されました.9.2; http://treesoft.sourceforge.net/treebest.shtml). 最後に、PAMLバージョン4.4ソフトウェアパッケージ42のMCMCTreeを使用して、八系統間の発散時間を推定しました。 化石記録に基づいて二つのプリオールは、ボレオウテリア(91-102MYA)とカルニボラ(52-57MYA)43を含む置換率を校正するために使用されました。 以前の研究と一致して、発見されたハイエナグループは、ネコ科(国内の犬とジャイアントパンダに代表される)53.9Mya44から分岐したネコ科の亜目を定義するクレードに含まれていた。

C.crocutaと6,601単一コピーオルソログに基づいて最尤法によって構築された七つの他の種の系統樹。 発散時間は、赤い菱形でマークされているタイムツリーデータベース(http://www.timetree.org)から導出された二つの校正事前値を使用して推定されました。 すべての推定発散時間は、95%信頼区間で括弧内に示されています。