페놀-클로로포름을 사용하여 게놈을 추출하고 에탄올 침전물을 사용하여 정제 하였다 13. 추출된 유전자는 1 에서 실행되고 시각화되었습니다.5%아가로오스 겔이 1 배 버퍼링되어 고분자량의 유전자가 있는지 확인합니다. 2000 년 나노드롭 2000 분광광도계 및 큐비트 2.0 형광계(미국 써모 피셔 사이언티픽)에서 농도 및 순도를 정량화하였다. 우리는 큐 비트 2.0 형광계를 사용하여 4 개의 복제 판독 값을 기반으로 나노 드롭 2000 및 0.245–0.399 를 사용하여 0.418 의 농도로 총 372 개의 게놈 유전자를 얻었습니다. 순도의 260/280 비율은 1.95 였다. 그런 다음 시토크롬 유전자를 사용하여 샘플을 바코드화했습니다. 그런 다음 그라디언트 라이브러리 전략에 따라 삽입 크기 길이가 170,500,800 인 13 개의 삽입 크기 라이브러리를 구성했습니다. 우리는 하이서크를 사용했습니다. 2000 시퀀서(일루미나,미국)쌍 끝을 시퀀싱하기(체육)14 레인에 걸쳐 각 라이브러리에 대한 읽습니다. 약 299 기가 바이트 원시 데이터의 총 149.25(표 1)의 시퀀싱 깊이(범위)를 달성,13 라이브러리로부터 생성되었다.
표 1 원시 읽기 데이터의 통계,게놈 크기를 가정하면 2.0 기가바이트입니다.
품질 관리
오조립 오류를 최소화하기 위해,우리는 다음 두 가지 기준에 따라 드 노보 게놈 어셈블리 이전에 원시 읽기를 필터링. 첫째,어댑터 시퀀스에 10 개 이상의 혈압이 정렬 된 읽기(<=3 혈압 불일치 허용)가 제거되었습니다. 둘째,10 보다 작거나 같은 품질 값을 갖는 기지의 40%가 삭제 된 읽습니다. 마지막으로,우리는 95.2 의 적용 범위를 가진 190.4 그램 데이터를 얻었다(표 2).
표 2 원시 읽기 데이터의 필터링에 따른 데이터 통계.
게놈 크기 추정
3 개의 짧은 삽입 라이브러리(170 혈압 중 2 개 및 500 혈압 중 하나)를 사용하여 게놈 크기 및 게놈 전체 이형 접합성을 추정했습니다. 해파리 14 에 대한 총 385 미터 판독 값이 제출되어 케이메르 주파수를 계산했습니다. 그런 다음 케이-메르 분포는 매개 변수와 게놈 스코프에 의해 설명되었다 7″케이=17;길이=100;최대 범위=1000″. 우리는 추정 된 게놈 크기 2,003,681,234 혈압과 0.325%의 이형 접합성을 얻었다(그림. 2).
그림. 2
17-게놈 크기의 메르 추정. 그만큼 엑스-축 이다 깊이(엑스),와이-축은 그 깊이의 주파수를 모든 커버리지 깊이의 총 주파수로 나눈 비율입니다. 서열 오류율,이형 접합률 및 게놈의 반복 속도를 고려하지 않고 17 메르 분포는 포아송 분포에 근접해야합니다.
유전체 조립 및 평가
06)15 는 짧은 삽입 크기 데이터의 필터링 및 큰 삽입 크기 데이터의 작은 피크를 제거한 다음 게놈 드 노보를 조립하기 위해 사용되었다. 소프 데노 보 어셈블리 알고리즘에는 세 가지 주요 단계가 포함되었습니다. (1)콘티그 구성:짧은 인서트 크기의 라이브러리 데이터를 케이메르로 분할하여 드 브루 인 그래프를 사용하여 구성했습니다.이 그래프는 팁 제거,거품 병합,연결의 낮은 커버리지 제거 및 작은 반복 제거로 단순화되었습니다. 그 결과 총 길이는 2,295,545,898 입니다. (2)비계 구조:우리는 콘티그에서 사용 가능한 모든 읽기를 재조정하여 모든 정렬 된 쌍 끝 읽기의 80%를 얻었습니다. 그런 다음 각 콘티그 쌍 사이의 공유 쌍 끝 관계의 양을 계산하고 일관된 쌍 끝과 충돌하는 쌍 끝의 비율을 가중시킨 다음 단계적으로 스캐 폴드를 구성했습니다. 그 결과,우리는 50 7,168,038 혈압,총 길이 2,355,303,269 혈압 짧은 삽입 크기 쌍 끝,긴 먼 쌍 끝에 비계를 얻을. (3)갭 닫기:건설 된 발판 내부의 간격을 채우기 위해,우리는 이러한 수집 읽기에 대한 로컬 어셈블리를 다시 할 읽기 쌍을 검색하는 쌍 엔드 정보를 사용. 요약하면,우리는 비계 내 갭의 87.7%또는 합계 갭 길이의 85.8%를 마감했습니다. 2015 년 11 월 23 일(토)부터 2015 년 12 월 31 일(일)까지,2015 년 12 월 31 일(일)까지,2015 년 12 월 31 일(일)까지. 스캐 폴드 어셈블리 크기는 2,355,303,269 혈압으로,스트라이프 하이에나,하이에나 하이에나에 대해보고 된 어셈블리 기반 게놈 크기 2,374,716,107 혈압에 가깝습니다. 우리는 또한 검색 하 고이 종에 대 한 염기 서열 첫 번째 미토콘드리아 게놈과 비슷한 16,858 혈압의 길이 있는 미토즈 프로그램을 사용 하 여 발견 하이에나의 미토콘드리아 게놈 주석 12.
표 3 조립된 시퀀스 길이의 통계.
게놈 초안의 평가는 부스코(버전 3.1.0)17 을 사용하여 단일 복사 직교체의 완전성을 조사함으로써 수행되었다. 오르 로그의 총 95.5%는 완전한 것으로,2.5%는 조각난 것으로,2.0%는 누락 된 것으로 확인되어 발견 된 하이에나 게놈 어셈블리의 전반적인 높은 품질을 나타냅니다. 짧은 비계(<1 천개)의 99.95%가 단지 1 개만을 숨겼다는 것을 감안할 때.총 게놈 길이의 2%,반복적인 요소와 유전자 기능 주석을 포함 하 여 다운스트림 분석에 대 한 이러한 발판 제외.
반복 요소 주석
탠덤 반복 및 전치 요소(테)에 대 한 검색 하 고 다 크로 쿠 타 게놈에 걸쳐 확인 했다. 탠덤 반복 찾기(재단,4.07)18 를 사용 하 여 확인 했다 및 전치 요소(테스)상 동성 기반 및 드 노 보 접근 방식의 조합에 의해 확인 되었다. 상 동성 기반 예측을 위해 반복 마스크 버전 4.0 을 사용했습니다.619 설정”-노로우-노로나-엔진 엔씨비”및 리피터틴마스크(리피터마스크 패키지 내의 프로그램)설정”-엔진 엔씨비-노로우-간단한-피값 0.0001″으로 알려진 반복에 기초하여 뉴클레오티드 및 아미노산 수준에서 테스를 검색한다(그림 1). 3). 이 라이브러리는 특정 실행 프로세스에서 불러오거나 실행될 수 있습니다 단백질 수준에서 반복 단백질 마스크를 사용하여 테 단백질 데이터베이스에 대해 링 블라스트를 수행했습니다. 이 예제에서는 다음과 같은 방법을 사용합니다.06)22 는 드 노보 반복 라이브러리를 구성하기 위해 적용되었다. 라이브러리의 오염 및 다중 복사 시퀀스를 제거하고 나머지 시퀀스는 스위스프로트 데이터베이스에 맞춰 폭발 결과에 따라 분류하였다. 이 라이브러리를 기반으로,우리는 상동 테스를 마스크 반복 마스크를 사용하고이를 분류(그림. 4). 전반적으로,반복적 인 요소의 826 메가바이트의 총 전체 게놈의 35.29%를 포함하는 발견 하이에나에서 확인되었다(표 4).
그림. 3
크로쿠타 크로쿠타 게놈 어셈블리 상동 기반 예측에 따라 전치 요소의 각 유형의 발산 속도의 분포. 발산 속도 상 동성 기반 메서드를 사용 하 여 게놈에서 확인 된 테스 및 렙베이스 데이터베이스 20 에서 합의 시퀀스 사이 계산 했다.
그림. 4
초기 예측에 기초한 크로쿠타 크로쿠타 게놈 어셈블리에서 각 유형의 테의 발산 속도의 분포. 발산 속도 순 초기 예측에 의해 게놈에서 확인 된 테와 예측된 테 라이브러리에서 합의 시퀀스 사이 계산 했다(방법 참조).
표 4 크로 투타 크로 투타 게놈 어셈블리의 전치 가능한 요소 함량.
단백질 코딩 유전자 주석
우리는 단백질 코딩 유전자뿐만 아니라 접합 사이트 및 대체 접합 이소 폼에 주석을 달기 위해 초기 예측 및 동종 기반 접근법을 사용했습니다. 25,스냅(버전 2006-07-28)26 을 각각 사용하여 인간,국내 개 및 국내 고양이의 유전자 모델을 사용하여 반복 마스크 된 게놈에 대한 예측을 수행 하였다. 이 방법으로 총 22,789 개의 유전자가 확인되었습니다. 상동 단백질,호모 사피엔스,펠리스 카투스 과 캐니스 익숙(앙상블 96 릴리스에서)을 사용하여 발견 된 하이에나 게놈에 매핑되었습니다. 그런 다음 정렬 된 서열과 쿼리 단백질은 정확한 접합 정렬을 검색하기 위해 유전자 별(버전 2.4.1)28 에 제출되었습니다. 최종 유전자 세트(22,747)는 맞춤형 파이프 라인을 사용하여 초기 및 동종 기반 결과를 병합하여 수집되었습니다(표 5).
표 5 에 기초한 단백질 코딩 유전자의 수에 대한 일반적인 통계 초기(드 노보)및 상 동성 기반 예측 방법.
유전자 기능 주석
유전자 기능 매개 변수를 사용 하 여 번역 된 유전자 코딩 서열을 정렬 하 여 얻은 최상의 일치에 따라 할당 되었습니다. 또한,유전자는 유전자에 의해 생성되고,유전자는 유전자에 의해 생성된다. 각 유전자에 대한 유전자 온톨로지 식별자는 해당 스위스프로트 및 트레블 항목으로부터 획득되었다. 모든 유전자는 케그 단백질에 대해 정렬되었으며 유전자가 관련 될 수있는 경로는 케그 데이터베이스 36 의 일치 된 유전자에서 파생되었습니다. 요약하면,예측 된 단백질 코딩 유전자의 22,166(97.45%)은 6 개의 데이터베이스 중 적어도 하나에 의해 성공적으로 주석을 달았습니다(표 6).
표 6 다른 단백질 데이터베이스에 대한 정렬에 따라 상 동성 또는 기능적 분류가 예측 된 유전자의 수.
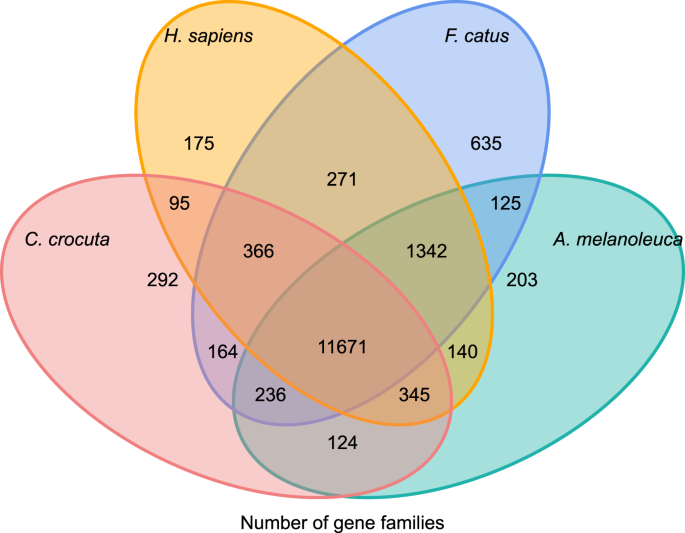
크로쿠타 크로쿠타의 유전자 계열의 계통 발생 역사와 진화에 대한 통찰력을 얻기 위해,우리는 7 종의 유전자 서열(펠리스카투스,카니스 익숙,아일 루로 포다 멜라노 루카,크로쿠타 크로쿠타,표범 속 파르 더스,표범 속 레오,표범 속 티그리스 알타이 카)과 호모 사피엔스를 아웃 그룹(앙상블 릴리스-96,미공개 데이터에서 표범 속 레오)으로 클러스터링했습니다.2015 년 11 월 1 일(토)~2015 년 11 월 1 일(일) 8 종에 대 한 단백질 코딩 유전자 다운스트림 쌍 할당(그래프 건물)에 대 한 각 유전자에 대 한 가장 긴 성적 증명서 등형을 선택 하 여 검색 했다. 우리는 모든 참조 종의 단백질 시퀀스에 대 한 모든 발파 검색을 수행,전자 값 컷오프 1 이자형-5. 유전자 가족 건설은’1.5’의 인플레이션 매개 변수를 사용하여 알고리즘 38 을 사용했습니다. 이 유전자는 유전자뿐만 아니라 유전자와도 관련이 있습니다. 이 네 종에 의해 공유 된 11,671 개의 유전자 패밀리가 있었고,1,446 개의 유전자를 포함하는 292 개의 유전자 패밀리는 크로 쿠타(그림 1)에 특이 적이었다. 5). 그 결과,사피엔스는 더 완전한 게놈과 주석을 가지고 있었다.
그림. 5
정형 분석을 기반으로 발견 하이에나,인간,국내 고양이 및 국내 개 사이에서 공유되고 독특한 단백질 코딩 유전자의 비교를 보여주는 벤 다이어그램.
우리는 8 종의 계통 발생 나무를 재구성 하는 6601 단일 복사 정형 유전자 확인. 각 유전자에 대한 아미노산 서열의 다중 서열 정렬은 근육(버전 3.8.31)39 를 사용하여 생성되었고,지블록(0.91 비)40 을 사용하여 트리밍되어,파라미터”-티=피-비 3=8-비 4=10-비 5=엔-이=-성”으로 잘 정렬된 영역을 달성하였다. 41 에서 구현 된 최대 우도 방법을 사용하여 계통 발생 분석을 수행했습니다. 6). 나무의 뿌리는 나무 베스트를 통해 전체 나무의 높이를 최소화함으로써 결정되었다(버전 1.9.2; http://treesoft.sourceforge.net/treebest.shtml). 최종적으로,우리는 팜플 버전 4.4 소프트웨어 패키지 42 에서 맥엠씨 트리를 사용하여 여덟 계통 중 발산 시간을 추정. 화석 기록을 기반으로 한 두 개의 선전이 보레 오우 테리아(91-102 미야)와 육식 동물(52-57 미야)43 을 포함하여 대체율을 보정하는 데 사용되었습니다. 이전 연구들과 일치하여,네 종의 고양이과에서 포함 된 발견 된 하이에나 그룹은 개과 동물(국내 개와 자이언트 팬더로 대표되는)에서 갈라진 아종 고양이 포모 미아를 정의하는 계통에 53.9 마이아 44.
그림. 6
크로쿠타의 계통 발생 나무와 6,601 개의 단일 복사 직교체를 기반으로 한 최대 우도 방법에 의해 구성된 7 개의 다른 종. 발산 시간은 빨간색 마름모로 표시된 시간 트리 데이터베이스(http://www.timetree.org)에서 파생 된 두 개의 교정 사전을 사용하여 추정되었습니다. 모든 예상 분기 시간은 괄호 안에 95%신뢰 구간으로 표시됩니다.