Vzorek kolekce, knihovna výstavby a sekvenování
Genomické DNA byla získána z mužského vzor C. crocuta (NCBI taxonomy ID: 9678; Obr. 1) uloženo ve Frozen Zoo® v San Diego Zoo Institute for Conservation Research, USA (ID zmrazené Zoo: KB4526).
genomová DNA byla extrahována za použití fenol-chloroformu a následným čištěním za použití vysrážení ethanolu13. Extrahovaná DNA byla spuštěna a vizualizována na 1.5% agarózový gel běží v 1x TBE pufru, aby se zkontrolovala přítomnost DNA s vysokou molekulovou hmotností. Koncentrace a čistota DNA byly kvantifikovány na spektrofotometru NanoDrop 2000 a Fluorometru Qubit 2.0 (Thermo Fisher Scientific, USA) před odesláním do BGI-Shenzhen, Čína. Získali jsme celkem 372 µg genomické DNA, s koncentrací 0.418 µg/µL pomocí Nanodrop 2000 a 0.245–0.399 µg/µL, založené na čtyři opakování měření pomocí Qubit 2.0 Fluorometer. Poměr čistoty 260/280 byl 1,95. Potom jsme čárový kód vzorku pomocí genu cytochromu b (Cytb). Poté jsme podle strategie gradient library zkonstruovali 13 knihoven velikosti vložky s následujícími délkami velikosti vložky: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Použili jsme HiSeq. 2000 sequencer (Illumina, USA) to sequence Paired-End (PE) čte pro každou knihovnu přes 14 pruhů. Celkem bylo vygenerováno asi 299 Gb nezpracovaných dat ze 13 knihoven, čímž bylo dosaženo hloubky sekvenování (pokrytí) 149,25 (Tabulka 1).
kontrola Kvality
minimalizovat misassembly chyby, jsme filtrované surové čte před de novo genomové montáž podle následujících dvou kritérií. Nejprve byly odstraněny čtení s více než 10 bp zarovnanými se sekvencí adaptéru (umožňující nesoulad <= 3 bp). Za druhé, čtení se 40% bází, které mají hodnotu kvality menší nebo rovnou 10, byly vyřazeny. Nakonec jsme získali 190,4 G data s pokrytím 95,2 (Tabulka 2).
Odhad velikosti genomu
Tři krátké-li vložit knihoven (dva 170 bp a jeden z 500 bp) byly použity pro odhad velikosti genomu a genomu-široký heterozygotnosti od k-mer analýzy. Celkem asi 385 M PE čtení bylo předloženo medúzy 14 pro výpočet frekvence k-mer. Pak byla distribuce k-mer ilustrována Genomescope7 s parametry „k = 17; délka = 100; maximální pokrytí = 1000“. Získali jsme odhadovanou velikost genomu 2,003,681,234 bp a heterozygotnost 0,325% (obr. 2).

17-mer odhad velikosti genomu. Osa x je hloubka (X), osa y je poměr, který představuje frekvenci v této hloubce děleno celkovou frekvencí všech hloubek pokrytí. Bez ohledu na rychlost chyb sekvence, rychlost heterozygotnosti a rychlost opakování genomu by distribuce 17-mer měla přiblížit poissonovu distribuci.
sestavení a hodnocení genomu
SOAPdenovo (V1.06) 15 byl použit k sestavení genomu de novo, po filtrování dat o velikosti krátké vložky a odstranění malého vrcholu dat o velikosti velké vložky. Algoritmus sestavy SOAPdenovo zahrnoval tři hlavní kroky. (1) konstrukce Contig: data knihovny velikosti krátké vložky byla rozdělena na k-mers a konstruována pomocí grafu de Bruijn, který byl zjednodušen odstraněním špiček, sloučením bublin, odstraněním nízkého pokrytí spojení a odstraněním malých opakování. Sekvenci contig jsme získali spojením dráhy k-mer, což mělo za následek kontig N50 2,104 bp a celkovou délku 2,295,545,898 bp. (2) konstrukce lešení: získali jsme 80% všech zarovnaných spárovaných koncových čtení přeskupením všech použitelných čtení na kontigách. Pak jsme vypočítali množství sdílené spárované-end vztahů mezi každý pár contigs, vážené sazby konzistentní a konfliktní spárované-končí, a pak postavené lešení krok za krokem. V důsledku toho jsme získali lešení s N50 7,168,038 bp a celkovou délkou 2,355,303,269 bp od krátkých párových konců velikosti vložky až po dlouhé vzdálené párové konce. (3) Mezera zavírání: zaplnit mezery uvnitř konstrukce lešení, jsme použili párový-end informace pro načtení číst párů dělat místní shromáždění znovu pro tyto shromážděné čte. V souhrnu jsme uzavřeli 87,7% mezer uvnitř lešení nebo 85,8% délky mezery součtu. Velikost contig N50 se zvýšila z 2 104 bp na 21 301 bp (Tabulka 3). Lešení montáž velikost byla 2,355,303,269 bp, což je blízko k sestavě-na základě velikosti genomu z 2,374,716,107 bp uvádí pro pruhované hyena, Hyena hyaena11 (NCBI přistoupení: ASM300989v1). Jsme také vyvolány a komentovaný mitochondriální genom hyena skvrnitá pomocí MitoZ program16, který má délku 16,858 bp, podobná první mitochondriální genom sekvenován pro tento species12.
Posouzení návrhu genomu byla provedena při pohledu na úplnost single-kopírování orthologs pomocí BUSCO (verze 3.1.0)17, vyhledávání proti Mammaliaodb9 databáze, která obsahuje 4,104 single-kopírování ortholog skupin. Celkem 95,5% ortologů bylo identifikováno jako úplné, 2,5% jako fragmentované a 2,0% jako chybějící, což svědčí o celkové vysoké kvalitě sestavy genomu hyeny skvrnité. Vzhledem k tomu, že 99,95% krátkých lešení (<1k) ukrývalo pouze 1.2% z celkové délky genomu jsme vyloučili tato lešení pro následnou analýzu, včetně anotace opakujících se prvků a genových funkcí.
anotace opakujících se prvků
oba tandemové opakování a transpozovatelné prvky (TE)byly hledány a identifikovány napříč genomem C. crocuta. Tandem se opakuje byly identifikovány pomocí Tandemové Opakuje Finder (TRF, v4.07)18 a transponovatelných elementů (TEs) byly identifikovány pomocí kombinace homologie-based a de novo přístupy. Pro predikci založenou na homologii jsme použili RepeatMasker verze 4.0.619 s nastavením „-nolow -no_is -norna -motor ncbi“ a RepeatProteinMask (v rámci programu RepeatMasker balíček) s nastavením „-motor ncbi -noLowSimple -phodnota 0.0001“ vyhledávání TEs v nukleotidové a aminokyselinové úrovni na základě známých opakuje (Obr. 3). RepeatMasker byl použit pro identifikaci na úrovni DNA pomocí vlastní knihovny, která kombinovala Dataset20 Repbase21. 10. Na úrovni proteinu byla použita RepeatProteinMask k provedení Rmblastu proti databázi TE proteinu. Pro ab initio predikce, RepeatModeler (v1. 0. 8) 21 a LTR_FINDING (v1.06) 22 byly použity ke konstrukci knihovny de novo repeat. Kontaminace a multi-copy sekvence v knihovně byly odstraněny a zbývající sekvence byly klasifikovány podle VÝBUCH výsledek po vyrovnání do databáze SwissProt. Na základě této knihovny jsme použili RepeatMasker k maskování homologních TEs a klasifikovali je (obr. 4). Celkově, celkem 826 Mb opakující se prvky, které byly identifikovány v hyena skvrnitá, zahrnující 35.29% z celého genomu (Tabulka 4).

Distribuce divergence rychlost každého typu transponovatelných elementů (TE), v Crocuta crocuta genomu shromáždění na základě homologie-na základě predikce. Míra divergence byla vypočtena mezi identifikovanými TEs v genomu pomocí metody založené na homologii a konsensuální sekvencí v databázi Repbase 20.

Distribuce divergence rychlost každého typu TE v Crocuta crocuta genomu shromáždění na základě ab initio predikce. Míra divergence byla vypočtena mezi identifikovanými TEs v genomu pomocí predikce ab initio a konsensuální sekvencí v předpovídané knihovně TE(viz metody).
Protein-kódujících genů anotace
Jsme použili ab initio predikce a homolog-based přístupy k anotaci protein-kódujících genů, stejně splétání sítě a alternativní sestřih izoformy. Ab initio predikce byla provedena na opakování-maskovaný genomu pomocí genové modely od člověka, pes domácí a kočka domácí použití AUGUSTUS (verze 2.5.5)23, GENSCAN24, GlimmerHMM (verze 3.0.4)25, a SNAP (verze 2006-07-28)26, resp. Touto metodou bylo identifikováno celkem 22 789 genů. Homologní proteiny, Homo sapiens, Felis catus a Canis familiaris (z Ensembl 96 vydání) byly mapovány do hyena skvrnitá genomu pomocí tblastn (Blastall 2.2.26)27 s parametry „-e 1e-5“. Zarovnané sekvence, stejně jako jejich dotazovací proteiny, byly poté odeslány do GeneWise (verze 2.4.1)28 pro vyhledávání přesného spliced alignment. Konečná sada genů (22 747) byla shromážděna sloučením výsledků založených na Ab initio a homolog pomocí přizpůsobeného potrubí (Tabulka 5).
Genové funkce anotace
Genové funkce byly přiřazeny podle nejlepší zápas, získané spojením přeloženou genů kódujících sekvencí pomocí BLASTP s parametry „-e 1e-5“ na SwissProt a TrEMBL databází (Uniprot vydání 2017-09). Motivy a domény genů byly určeny InterProScan (v5)29 proti proteinu databází včetně ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 a PROSITE35. ID ontologie genů pro každý gen byly získány z odpovídajících záznamů SwissProt a TrEMBL. Všechny geny byly zarovnány s proteiny KEGG a cesta, do které by mohl být gen zapojen, byla odvozena z odpovídajících genů v databázi kegg36. V souhrnu, 22,166 (97.45%) předpověděl protein-kódujících genů byly úspěšně komentovaný tím, že alespoň jeden ze šesti databází (Tabulka 6).
Genové rodiny výstavba a rekonstrukce fylogeneze
získat vhled do fylogenetické historii a evoluci genových rodin Crocuta crocuta, jsme shlukli genových sekvencí sedmi druhů (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) a Homo sapiens jako outgroup (Ensembl release-96, Panthera leo z nepublikované údaje) do genové rodiny pomocí orthoMCL (v2.0.9)37. Geny kódující proteiny pro osm druhů byly získány výběrem nejdelší izoformy transkriptu pro každý gen pro následné párové přiřazení(vytváření grafů). Provedli jsme vše proti všem BLASTP vyhledávání proteinových sekvencí všech referenčních druhů, s mezní hodnotou E 1e-5. Konstrukce genové rodiny používala algoritmus MCL38 s inflačním parametrem „1.5“. Celkem 16,271 genové rodiny C. crocuta, H. sapiens, F. catus, A. melanoleuca shlukli. Tam byly 11,671 genových rodin sdílí tyto čtyři druhy, zatímco 292 genové rodiny obsahují 1,446 geny jsou specifické pro C. Crocuta (Obr. 5). Výrazně, genové rodiny C. crocuta a F. catus sdílené méně než C. crocuta a H. sapiens sdílené, které může mít za následek, že H. sapiens měl více kompletní genom a anotace.
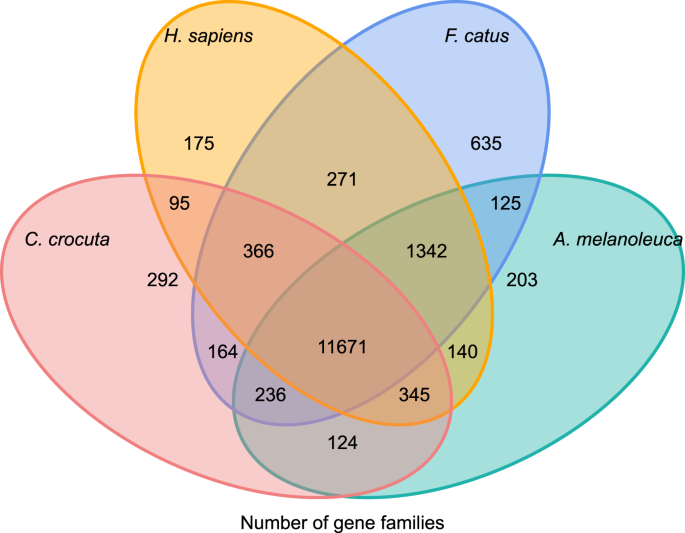
Vennův diagram zobrazující srovnání společných a jedinečný protein-kódujících genů mezi hyena skvrnitá, člověk, kočka domácí a pes domácí založené na orthology analýzy.
identifikovali Jsme 6,601 single-kopírování orthologous geny pro rekonstrukci fylogenetických stromů z osmi druhů. Více sekvence zarovnání sekvencí aminokyselin pro každý gen byly generovány pomocí SVALŮ (verze 3.8.31)39, a zdobené pomocí Gblocks (0.91 b)40, dosažení dobře srovnané regiony s parametry „-t = p -b3 = 8 -b4 = 10 -b5 = n -e = -st“. Provedli jsme fylogenetickou analýzu metodou maximální pravděpodobnosti implementovanou v PhyML (v3.0)41, pomocí modelu JTT + G + I pro substituci aminokyselin (obr. 6). Kořen stromu byl určen minimalizací výšky celého stromu pomocí Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Nakonec jsme odhadli čas divergence mezi osmi liniemi pomocí MCMCTree ze softwarového balíčku PAML verze 4.4. Ke kalibraci míry substituce byly použity dva převory založené na fosilním záznamu, včetně Boreoeutheria (91-102 MYA) a Carnivora (52-57 MYA)43. V souladu s předchozími studiemi, hyena skvrnitá skupin s čtyři druhů v ceně od kočkovitých šelem v clade definování podřádu Feliformia, který se odchyloval od Caniformia (zastoupené domácí pes a panda) 53.9 Mya44.

Fylogenetický strom pro C crocuta a sedm dalších druhů postavené metoda maximální věrohodnosti na základě 6,601 single-kopírování orthologues. Doba divergence byla odhadnuta pomocí dvou kalibračních priors odvozených z databáze časového stromu (http://www.timetree.org), které jsou označeny červeným kosočtvercem. Všechny odhadované časy divergence jsou uvedeny s 95% intervaly spolehlivosti v závorkách.