Spinální Muskulární Atrofie
Stáhněte si náš Spinální Muskulární Atrofie List
Dozvědět se o MDA je COVID-19 reakce
Co je to spinální svalová atrofie?
spinální svalová atrofie (SMA) je genetické onemocnění postihující centrální nervový systém, periferní nervový systém a dobrovolný pohyb svalů (kosterní sval).
většina nervových buněk, které kontrolují svaly, se nachází v míše, což představuje slovo spinální ve jménu nemoci. SMA je svalová, protože její primární účinek je na svaly, které nepřijímají signály z těchto nervových buněk. Atrofie je lékařský termín pro stále menší, což je to, co se obvykle stane se svaly, když nejsou stimulovány nervovými buňkami.
SMA zahrnuje ztrátu nervových buněk nazývaných motorické neurony v míše a je klasifikována jako onemocnění motorických neuronů.
v nejběžnější formě SMA (chromozom 5 SMA nebo SMA související s SMN) existuje široká variabilita věku nástupu, symptomů a rychlosti progrese. Za účelem zohlednění těchto rozdílů je SMA související s chromozomem 5, který je často autozomálně recesivní,klasifikován do typů 1 až 4.
věk, ve kterém SMA příznaky začínají zhruba koreluje s mírou, do které motorické funkce je ovlivněna: dřívější věk nástupu, tím větší dopad na motorické funkce. Děti, které vykazují příznaky při narození nebo v dětství, mají obvykle nejnižší úroveň fungování (typ 1). Pozdější nástup SMA s méně závažným průběhem (typy 2 a 3 a u dospívajících nebo dospělých typ 4) obecně koreluje se stále vyššími úrovněmi motorických funkcí.
více viz formuláře SMA.
co způsobuje SMA?
chromozom 5 SMA je způsoben nedostatkem proteinu motorického neuronu zvaného SMN, pro “ přežití motorického neuronu.“Tento protein, jak naznačuje jeho název, se zdá být nezbytný pro normální funkci motorických neuronů. SMN hraje klíčovou roli v genové expresi v motorických neuronech. Jeho nedostatek je způsoben genetickými vadami (mutacemi) na chromozomu 5 v genu zvaném SMN1. Nejčastější mutace v SMN1 genu v rámci pacientů s diagnózou SMA je vypuštění celého segmentu, tzv. exon 7.1 Sousední SMN2 genů lze částečně kompenzovat nefunkční geny SMN1, jak tam je 99% identity mezi těmito dvěma geny.2
Další vzácné formy SMA (non-chromozomu 5) jsou způsobeny mutacemi v genech jiných než SMN1.3
Pro více informací, včetně podrobného čtení na vzácné, non-chromozomu 5-linked SMA, viz Formy SMA a Příčiny/Dědictví.
jaké jsou příznaky SMA?
příznaky SMA pokrývají široké spektrum, od mírných po těžké.
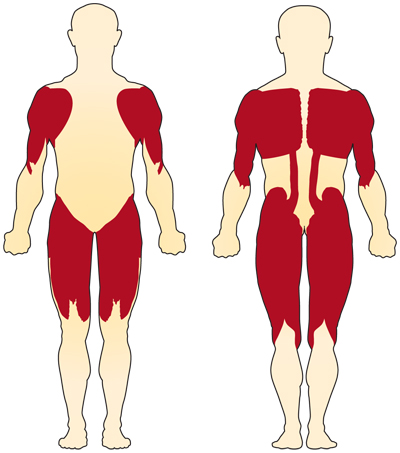
primárním příznakem SMA související s chromozomem 5 (související s SMN) je slabost dobrovolných svalů. Nejvíce postižené svaly jsou ty nejblíže ke středu těla, jako jsou svaly ramen, boků, stehen a horní části zad. Zdá se, že dolní končetiny jsou postiženy více než horní končetiny a hluboké reflexy šlach jsou sníženy.4
zvláštní komplikace se vyskytují, pokud jsou postiženy svaly používané k dýchání a polykání, což má za následek abnormality v těchto funkcích. Pokud svaly na zádech oslabí, mohou se vyvinout zakřivení páteře.
existuje velké množství variací ve věku nástupu a úrovni motorické funkce dosažené v SMA související s chromozomem 5. Ty zhruba korelují s tím, kolik funkčního proteinu SMN je přítomno v motorických neuronech, což zase koreluje s tím, kolik kopií genů SMN2 má člověk. Smyslové, mentální a emoční fungování jsou v chromozomu-5 SMA zcela normální.
některé formy SMA nejsou spojeny s nedostatkem chromozomu 5 nebo SMN. Tyto formy se velmi liší v závažnosti a ve svalech nejvíce postižených. Zatímco většina forem, jako chromozomu 5-související formě, vliv především na proximální svaly, jiné formy existují, které ovlivňují hlavně distální svaly (ty dál od těla centra) — alespoň na začátku.
více viz příznaky a symptomy.
jaký je postup SMA?
v SMA související s chromozomem 5, čím později začnou příznaky a čím více proteinu SMN je, tím mírnější bude průběh onemocnění. Zatímco v minulosti, dětí s SMA obvykle nepřežila více než dva roky, dnes většina lékařů se nyní uvažovat SMN související s SMA být kontinua a nechcete, aby se tuhý předpovědi o délku života, nebo slabost, založené výhradně na věk nástupu.
SMA je nejčastější genetickou příčinou úmrtnosti u kojenců.
jaký je stav výzkumu SMA?
výzkum se zaměřil na strategie ke zvýšení produkce proteinu SMN v těle, který chybí ve formách onemocnění souvisejících s chromozomem 5. Přístupy zahrnují metody, které pomáhají motorickým neuronům přežít za nepříznivých okolností.
Dne Dec. 23, 2016, americký Úřad pro kontrolu potravin a léčiv (FDA) schválil Spinraza (nusinersen) pro léčbu SMA. Přípravek Spinraza je určen k léčbě základní vady SMA, což znamená, že může být účinný při zpomalení, zastavení nebo možná zvrácení příznaků SMA. Více viz Spinraza je schválen.
v květnu 2019 FDA schválila ZOLGENSMA (onasemnogen abeparvovac-xioi), první genovou substituční terapii neuromuskulárního onemocnění. Zolgensma je jednorázová intravenózní (do žíly) infuze pro léčbu pediatrických pacientů mladších než 2 roky věku s SMA s bi-allelickou mutace v SMN1 genu, včetně těch, kteří jsou presymptomatickými na diagnostiku. Pro více informací si přečtěte FDA schvaluje přípravek AveXis Zolgensma pro léčbu spinální svalové atrofie u dětských pacientů.
více viz výzkum, SMA: plná rychlost vpřed a v centru pozornosti: spinální svalová atrofie (SMA). Pro příběhy rodin žijících s SMA, podívejte se na naše příběhy SMA silně, blog MDA.
V srpnu roku 2020, FDA schválila risdiplam (značka Evrysdi*) pro léčbu SMA u dospělých a dětí od dvou měsíců věku nebo starší. Evysdi je perorální lék určený ke zvýšení hladiny proteinu SMN zvýšením produkce genu SMN2 „backup“.
- Ogino, s. & Wilson, R. B. genetické testování a hodnocení rizik spinální svalové atrofie (SMA). Lidská Genetika (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identifikace a charakterizace genu určujícího spinální svalovou atrofii. Buňka (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. Non-5q spinální svalové atrofie: alfanumerická polévka zhoustne. Neurologie (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & kissel, J. T. spinální svalová atrofie: diagnostika a léčba v nové terapeutické éře. Svaly a nervy (2015). doi: 10.1002 / mus.24497