Probensammlung, Bibliothekskonstruktion und Sequenzierung
Genomische DNA wurde von einem männlichen Exemplar von C. crocuta erhalten (NCBI Taxonomie ID: 9678; Abb. 1) gelagert im Frozen Zoo® am San Diego Zoo Institute for Conservation Research, USA (Frozen Zoo ID: KB4526).
Die genomische DNA wurde mit Phenol-Chloroform extrahiert, gefolgt von einer Reinigung mit Ethanolfällung13. Die extrahierte DNA wurde ausgeführt und auf einem visualisiert 1.5% Agarosegel in 1x TBE-Puffer laufen lassen, um das Vorhandensein von DNA mit hohem Molekulargewicht zu überprüfen. DNA-Konzentration und Reinheit wurden auf einem NanoDrop 2000 Spektralphotometer und Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA) vor dem Versand nach BGI-Shenzhen, China quantifiziert. Wir erhielten insgesamt 372 µg genomische DNA mit einer Konzentration von 0,418 µg / µL unter Verwendung des Nanodrop 2000 und 0,245–0,399 µg / µL basierend auf vier Replikatmessungen unter Verwendung des Qubit 2.0-Fluorometers. Das Reinheitsverhältnis 260/280 betrug 1,95. Wir haben dann die Probe mit dem Cytochrom b (Cytb) -Gen barcodiert. Dann haben wir gemäß der Gradientenbibliotheksstrategie 13 Bibliotheken mit Einfügegröße mit den folgenden Einfügegrößenlängen erstellt: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Wir haben den HiSeq benutzt. 2000 Sequenzer (Illumina, USA) zur Sequenzierung von Paired-End (PE)-Lesevorgängen für jede Bibliothek auf 14 Spuren. Insgesamt wurden aus 13 Bibliotheken ca.299 GB Rohdaten generiert, die eine Sequenziertiefe (Coverage) von 149,25 erreichten (Tabelle 1).
Qualitätskontrolle
Um Fehler bei der Montage zu minimieren, haben wir die Rohdaten vor der de novo-Genommontage nach den folgenden zwei Kriterien gefiltert. Zuerst wurden Lesevorgänge mit mehr als 10 bp, die auf die Adaptersequenz ausgerichtet waren (was eine Fehlanpassung von < = 3 bp zuließ), entfernt. Zweitens wurden Lesevorgänge mit 40% der Basen mit einem Qualitätswert kleiner oder gleich 10 verworfen. Schließlich erhielten wir 190,4 G Daten mit einer Abdeckung von 95,2 (Tabelle 2).
Schätzung der Genomgröße
Drei Short-Insert-Bibliotheken (zwei mit 170 bp und eine mit 500 bp) wurden verwendet, um die Genomgröße und die genomweite Heterozygotie durch k-mer-Analyse abzuschätzen. Insgesamt wurden etwa 385 M PE-Reads an jellyfish14 gesendet, um die k-Mer-Frequenz zu berechnen. Anschließend wurde die k-mer-Verteilung durch Genomescope7 mit den Parametern „k = 17; length = 100; max coverage = 1000“ dargestellt. Wir erhielten eine geschätzte Genomgröße von 2.003.681.234 bp und eine Heterozygotie von 0,325% (Abb. 2).

17- mer Schätzung der Genomgröße. Die x-Achse ist die Tiefe (X), die y-Achse ist der Anteil, der die Frequenz in dieser Tiefe dividiert durch die Gesamtfrequenz aller Erfassungstiefen darstellt. Ohne Berücksichtigung der Sequenzfehlerrate, der Heterozygotätsrate und der Wiederholungsrate des Genoms sollte sich die 17-mer-Verteilung einer Poisson-Verteilung annähern.
Genom Montage und Bewertung
SOAPdenovo (V1.06) 15 wurde verwendet, um das Genom de novo zusammenzusetzen, nach dem Filtern der kurzen Insertgrößendaten und Entfernen des kleinen Peaks der großen Insertgrößendaten. Der SOAPdenovo-Montagealgorithmus umfasste drei Hauptschritte. (1) Zusammenhängende Konstruktion: Die Bibliotheksdaten mit kurzer Einfügegröße wurden in k-mers aufgeteilt und unter Verwendung eines de Bruijn-Diagramms erstellt, das durch Entfernen von Spitzen, Zusammenführen von Blasen, Entfernen der geringen Abdeckung der Verbindung und Entfernen kleiner Wiederholungen vereinfacht wurde. Wir erhielten die Contig-Sequenz, indem wir den k-Mer-Pfad verbanden, was zu einem contig N50 2,104 bp und einer Gesamtlänge von 2,295,545,898 bp führte. (2) Gerüstbau: Wir haben 80% aller ausgerichteten Lesevorgänge am gepaarten Ende erhalten, indem wir alle verwendbaren Lesevorgänge an Contigs neu ausgerichtet haben. Dann berechneten wir die Anzahl der gemeinsamen Paired-End-Beziehungen zwischen jedem Paar von Contigs, gewichteten die Rate konsistenter und widersprüchlicher Paired-Ends und konstruierten dann die Gerüste Schritt für Schritt. Als Ergebnis erhielten wir Gerüste mit einem N50 von 7.168.038 bp und einer Gesamtlänge von 2.355.303.269 bp von kurzen gepaarten Enden in Einsatzgröße zu langen entfernten gepaarten Enden. (3) Lückenschluss: Um die Lücken in den konstruierten Gerüsten zu füllen, haben wir die gepaarten Endinformationen verwendet, um die Lesepaare abzurufen und für diese gesammelten Lesevorgänge erneut eine lokale Assembly durchzuführen. Zusammenfassend haben wir 87,7% der Intra-Scaffold-Lücken oder 85,8% der Summenspaltlänge geschlossen. Die zusammenhängende N50-Größe erhöhte sich von 2.104 bp auf 21.301 bp (Tabelle 3). Die Größe der Gerüstbaugruppe betrug 2.355.303.269 bp, was nahe an der assemblationsbasierten Genomgröße von 2.374.716.107 bp liegt, die für die gestreifte Hyäne Hyaena hyaena11 (NCBI accession: ASM300989v1) gemeldet wurde. Wir haben auch das mitochondriale Genom der gefleckten Hyäne mit dem MitoZ-programm16, das eine Länge von 16.858 bp hat, ähnlich den ersten mitochondrialen Genomen, die für diese Art sequenziert wurden12.
Die Bewertung des Entwurfsgenoms wurde durchgeführt, indem die Vollständigkeit von Einzelkopie-Orthologen unter Verwendung von BUSCO (Version 3.1.0) 17 untersucht und in der Mammaliaodb9-Datenbank gesucht wurde, die 4.104 Einzelkopie-Ortholog-Gruppen enthält. Insgesamt wurden 95,5% der Orthologen als vollständig, 2,5% als fragmentiert und 2,0% als fehlend identifiziert, was auf eine insgesamt hohe Qualität der Genomassemblierung der gefleckten Hyänen hinweist. Da 99,95% der kurzen Gerüste (< 1k) nur 1 beherbergten.2% der gesamten Genomlänge haben wir diese Gerüste für die nachgelagerte Analyse ausgeschlossen, einschließlich der Annotation von repetitiven Elementen und Genmerkmalen.
Repetitive Element Annotation
Sowohl Tandem-Repeats als auch transposable Elemente (TE) wurden im gesamten C. crocuta-Genom gesucht und identifiziert. Tandem-Wiederholungen wurden mit dem Tandem Repeats Finder (TRF, v4.07)18 identifiziert und transposable Elemente (TEs) wurden durch eine Kombination von homologiebasierten und De-Novo-Ansätzen identifiziert. Für die homologiebasierte Vorhersage haben wir RepeatMasker Version 4.0 verwendet.619 mit den Einstellungen „-nolow -no_is -norna -engine ncbi“ und RepeatProteinMask (ein Programm innerhalb des RepeatMasker-Pakets) mit den Einstellungen „-engine ncbi -noLowSimple -pvalue 0.0001“, um TEs auf Nukleotid- und Aminosäurenebene basierend auf bekannten Wiederholungen zu suchen (Abb. 3). RepeatMasker wurde für die Identifizierung auf DNA-Ebene unter Verwendung einer benutzerdefinierten Bibliothek angewendet, die den Repbase21.10-Datensatz kombiniert20. Auf Proteinebene wurde RepeatProteinMask verwendet, um RMBlast gegen die TE-Proteindatenbank durchzuführen. Für ab initio Vorhersage, RepeatModeler (v1.0.8)21 und LTR_FINDING (v1.06)22 wurden angewendet, um die de novo repeat Library zu konstruieren. Kontaminations- und Multikopie-Sequenzen in der Bibliothek wurden entfernt und die restlichen Sequenzen nach Abgleich mit der SwissProt-Datenbank nach dem Blastergebnis klassifiziert. Basierend auf dieser Bibliothek haben wir RepeatMasker verwendet, um die homologen TEs zu maskieren und zu klassifizieren (Abb. 4). Insgesamt wurden in der gefleckten Hyäne 826 Mb repetitive Elemente identifiziert, die 35,29% des gesamten Genoms ausmachen (Tabelle 4).

Verteilung der Divergenzrate jeder Art von transposablem Element (TE) in der Crocuta Crocuta-Genomassemblierung basierend auf homologiebasierter Vorhersage. Die Divergenzrate wurde zwischen den identifizierten TEs im Genom unter Verwendung einer homologiebasierten Methode und der Konsensussequenz in der Repbase-Datenbank berechnet20.

Verteilung der Divergenzrate jeder Art von TE in der Crocuta crocuta Genomanordnung basierend auf ab initio Vorhersage. Die Divergenzrate wurde zwischen den identifizierten TEs im Genom durch Ab-initio-Vorhersage und der Konsensussequenz in der vorhergesagten TE-Bibliothek berechnet (siehe Methoden).
Protein-kodierende Genannotation
Wir verwendeten Ab Initio-Vorhersage und Homolog-basierte Ansätze, um Protein-kodierende Gene sowie Spleißstellen und alternative Spleißisoformen zu kommentieren. Die Ab-initio-Vorhersage wurde am wiederholungsmaskierten Genom unter Verwendung von Genmodellen von Mensch, Haushund und Hauskatze unter Verwendung von AUGUSTUS (Version 2.5.5) 23, GENSCAN24, GlimmerHMM (Version 3.0.4) 25 bzw. SNAP (Version 2006-07-28) 26 durchgeführt. Insgesamt wurden mit dieser Methode 22.789 Gene identifiziert. Homologe Proteine von Homo sapiens, Felis catus und Canis familiaris (aus der Ensembl 96-Veröffentlichung) wurden mit tblastn (Blastall 2.2.26)27 mit den Parametern „-e 1e-5“ auf das gefleckte Hyänengenom abgebildet. Die ausgerichteten Sequenzen sowie ihre Abfrageproteine wurden dann an GeneWise (Version 2.4.1) 28 zur Suche nach einer genauen gespleißten Ausrichtung gesendet. Der endgültige Gensatz (22.747) wurde durch Zusammenführen von Ab initio- und Homolog-basierten Ergebnissen unter Verwendung einer angepassten Pipeline gesammelt (Tabelle 5).
Annotation der Genfunktionen
Die Zuordnung der Genfunktionen erfolgte nach der besten Übereinstimmung, die durch Ausrichtung der translatierten genkodierenden Sequenzen mittels BLASTP mit den Parametern „-e 1e-5“ auf die Datenbanken SwissProt und TrEMBL (Uniprot Release 2017-09) erzielt wurde. Die Motive und Domänen von Genen wurden durch InterProScan (v5) 29 gegen Proteindatenbanken einschließlich ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 und PROSITE35 bestimmt. Genontologie-IDs für jedes Gen wurden aus den entsprechenden SwissProt- und TrEMBL-Einträgen erhalten. Alle Gene wurden gegen KEGG-Proteine ausgerichtet, und der Weg, an dem das Gen beteiligt sein könnte, wurde aus den übereinstimmenden Genen in der KEGG-Datenbank abgeleitet36. Zusammenfassend wurden 22.166 (97,45%) der vorhergesagten proteinkodierenden Gene von mindestens einer der sechs Datenbanken erfolgreich annotiert (Tabelle 6).
Genfamilienkonstruktion und Phylogenie-Rekonstruktion
Um einen Einblick in die phylogenetische Geschichte und Evolution von Genfamilien von Crocuta crocuta zu erhalten, gruppierten wir Gensequenzen von sieben Arten (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) und Homo sapiens als Outgroup (Ensembl Release-96, Panthera leo aus unveröffentlichten Daten) in Genfamilien unter Verwendung von orthoMCL (v2.0.9)37. Die Protein-kodierenden Gene für die acht Spezies wurden durch Auswahl der längsten Transkript-Isoform für jedes Gen für die paarweise Zuordnung nachgeschaltet (Graphenaufbau). Wir führten eine All-gegen-All-BLASTP-Suche an den Proteinsequenzen aller Referenzspezies mit einem E-Wert-Cut-off von 1e-5 durch. Die Genfamilienkonstruktion verwendete den MCL-Algorithmus38 mit dem Inflationsparameter ‚1,5‘. Insgesamt wurden 16.271 Genfamilien von C. crocuta, H. sapiens, F. catus, A. melanoleuca geclustert. Es gab 11.671 Genfamilien, die von diesen vier Arten geteilt wurden, während 292 Genfamilien mit 1.446 Genen spezifisch für C. Crocuta waren (Abb. 5). Auffallend ist, dass die Genfamilien C. crocuta und F. catus weniger gemeinsam hatten als C. crocuta und H. sapiens, was darauf zurückzuführen sein könnte, dass H. sapiens ein vollständigeres Genom und eine vollständigere Annotation hatte.
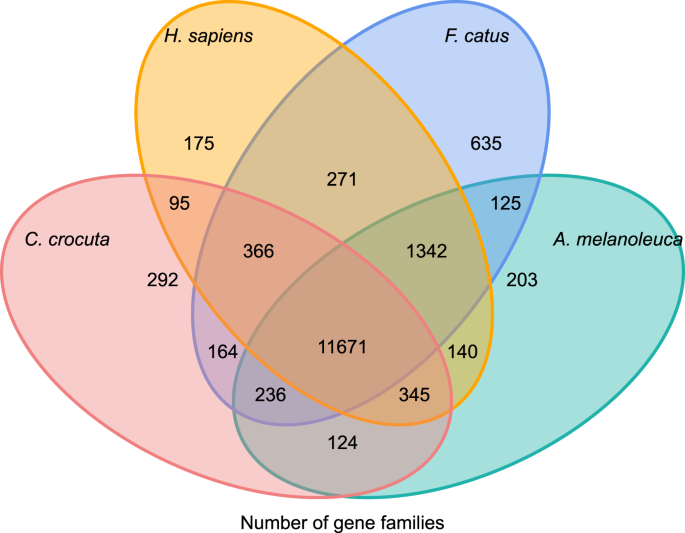
Venn-Diagramm, das den Vergleich gemeinsamer und einzigartiger proteinkodierender Gene zwischen gefleckter Hyäne zeigt, Mensch, Hauskatze und Haushund basierend auf Orthologieanalyse.
Wir identifizierten 6.601 orthologe Gene in Einzelkopie, um den phylogenetischen Baum der acht Arten zu rekonstruieren. Mehrere Sequenzausrichtungen von Aminosäuresequenzen für jedes Gen wurden unter Verwendung von MUSCLE (Version 3.8.31) 39 erzeugt und unter Verwendung von Gblocks (0.91b) 40 getrimmt, wobei gut ausgerichtete Regionen mit den Parametern „-t = p -b3 = 8 -b4 = 10 -b5 = n -e = -st“ erreicht wurden. Wir führten eine phylogenetische Analyse mit der Maximum-Likelihood-Methode durch, wie sie in PhyML (v3.0) 41 implementiert ist, unter Verwendung des JTT + G + I-Modells für die Aminosäuresubstitution (Abb. 6). Die Wurzel des Baumes wurde bestimmt, indem die Höhe des gesamten Baumes über Treebest (v1) minimiert wurde.9.2; http://treesoft.sourceforge.net/treebest.shtml). Schließlich schätzten wir die Divergenzzeit zwischen den acht Linien mit MCMCTree aus dem PAML-Softwarepaket Version 4.442. Zwei auf dem Fossilienbestand basierende Prioren wurden verwendet, um die Substitutionsrate zu kalibrieren, einschließlich Boreoeutheria (91-102 MYA) und Carnivora (52-57 MYA)43. In Übereinstimmung mit früheren Studien wurden die gefleckten Hyänengruppen mit den vier Arten aus den Felidae in einer Klade eingeschlossen, die die Unterordnung Feliformia definiert, die von der Caniformia (vertreten durch den Haushund und den Riesenpanda) 53.9 Mya44 abweicht.

Phylogenetischer Baum von C. crocuta und sieben anderen Arten, konstruiert nach der Maximum-Likelihood-Methode basierend auf 6.601 Einzelkopien-Orthologen. Die Divergenzzeit wurde unter Verwendung der beiden aus der Zeitbaumdatenbank abgeleiteten Kalibrierprioren (http://www.timetree.org) geschätzt, die durch eine rote Raute gekennzeichnet sind. Alle geschätzten Divergenzzeiten sind mit 95% -Konfidenzintervallen in Klammern angegeben.