Spinal Muscular Atrofy
Download our Spinal Muscular Atrofy Fact Sheet
Learn about MDA ’ s COVID-19 response
What is spinal muscular atrofy?
Spinal muscular atrofy (SMA) on keskushermostoon, ääreishermostoon ja vapaaehtoiseen lihasliikkeeseen (luustolihas) vaikuttava geneettinen sairaus.
suurin osa lihaksia säätelevistä hermosoluista sijaitsee selkäytimessä, mistä johtuu taudin nimessä oleva sana spinal. SMA on lihaksikas, koska sen ensisijainen vaikutus on lihaksissa, jotka eivät saa signaaleja näistä hermosoluista. Atrofia on lääketieteellinen termi pienenemiselle, joka yleensä tapahtuu lihaksille, kun hermosolut eivät stimuloi niitä.
SMA: ssa on kyse motoneuroneiksi kutsuttujen hermosolujen katoamisesta selkäytimestä ja se luokitellaan motoneuronitaudiksi.
yleisimmässä SMA: n muodossa (kromosomi 5 SMA eli SMN: ään liittyvä SMA) alkamisiässä, oireissa ja etenemisnopeudessa on suurta vaihtelua. Näiden erojen selittämiseksi kromosomiin 5 liittyvä SMA, joka on usein autosomaalisesti resessiivinen, luokitellaan tyyppeihin 1 – 4.
ikä, jolloin SMA-oireet alkavat, korreloi suurin piirtein siihen, missä määrin ne vaikuttavat motorisiin toimintoihin: mitä varhaisemmassa iässä ne ovat alkaneet, sitä suurempi vaikutus niillä on motorisiin toimintoihin. Lapsilla, joilla ilmenee oireita syntyessään tai vauvaiässä, on tyypillisesti alhaisin toimintakyky (tyyppi 1). Myöhemmin ilmenevä SMA, jonka kuuri on lievempi (tyypit 2 ja 3 ja teini-ikäisillä tai aikuisilla tyyppi 4), korreloi yleensä yhä suurempaan motoriseen toimintakykyyn.
Katso lisää SMA: n muotoja.
mikä aiheuttaa SMA: n?
kromosomi 5 SMA: n aiheuttaa SMN-nimisen motoneuroniproteiinin puutos, ”survival of motor neuron.”Tämä proteiini näyttää nimensä mukaisesti olevan välttämätön normaalille motoneuronitoiminnalle. SMN: llä on keskeinen rooli geenien ilmentymisessä motorisissa neuroneissa. Sen puutos johtuu geenivirheistä (mutaatioista) kromosomissa 5 geenissä nimeltä SMN1. Yleisin mutaatio smn1-geenissä potilailla, joilla on diagnosoitu SMA, on kokonaisen segmentin poisto, jota kutsutaan Exon 7.1-Viereisiksi Smn2-geeneiksi, voi osittain kompensoida toimimattomia SMN1-geenejä, koska näiden kahden geenin välillä on 99% identiteetti.2
muut harvinaiset SMA: n muodot (ei-kromosomi 5) johtuvat mutaatioista muissa geeneissä kuin SMN1.3
lisätietoja, mukaan lukien yksityiskohtainen lukeminen harvinaisesta, ei-kromosomiin 5 liittyvästä SMA: sta, KS.SMA: n muodot ja syyt/periytyminen.
mitä oireita SMA: lla on?
SMA-oireet kattavat laajan kirjon, joka vaihtelee lievästä vaikeaan.
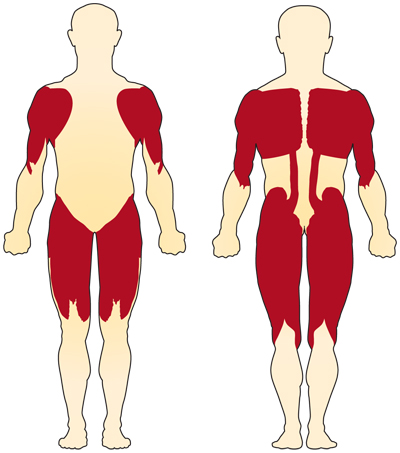
kromosomiin 5 liittyvän (SMN: ään liittyvän) SMA: n ensisijainen oire on vapaaehtoisten lihasten heikkous. Eniten se vaikuttaa lihaksiin, jotka ovat lähimpänä kehon keskustaa, kuten olkapäät, lonkat, reidet ja yläselkä. Alaraajat näyttävät kärsivän enemmän kuin yläraajat, ja syvät jännerefleksit heikkenevät.4
erityiskomplikaatioita esiintyy, jos hengitys-ja nielemishäiriöt aiheuttavat poikkeavuuksia näissä toiminnoissa. Jos selän lihakset heikkenevät, selkärangan kurvit voivat kehittyä.
kromosomiin 5 liittyvässä SMA: ssa on paljon vaihtelua alkamisiässä ja motorisen toiminnan tasossa. Nämä korreloivat suurin piirtein sen kanssa, kuinka paljon motoneuroneissa on funktionaalista SMN-proteiinia, mikä puolestaan korreloi sen kanssa, kuinka monta kopiota smn2-geeneistä ihmisellä on. Aistien, mielen ja tunteiden toiminta on täysin normaalia kromosomissa-5 SMA.
jotkin SMA: n muodot eivät liity kromosomi 5: n tai SMN: n puutokseen. Nämä muodot vaihtelevat suuresti vakavuudeltaan ja eniten kärsivissä lihaksissa. Vaikka useimmat muodot, kuten kromosomiin 5 liittyvä muoto, vaikuttavat enimmäkseen proksimaalisiin lihaksiin, on olemassa muita muotoja, jotka vaikuttavat enimmäkseen distaalisiin lihaksiin (ne, jotka ovat kauempana kehon keskustasta) — ainakin alussa.
lisää, Katso löydökset ja oireet.
miten SMA etenee?
kromosomiin 5 liittyvässä SMA: ssa taudin kulku on todennäköisesti sitä lievempi, mitä myöhemmin oireet alkavat ja mitä enemmän SMN-proteiinia on. Vaikka aiemmin SMA: ta sairastavat lapset eivät yleensä selvinneet yli kahta vuotta, nykyään useimmat lääkärit pitävät SMN: ään liittyvää SMA: ta jatkumona eivätkä halua tehdä jäykkiä ennusteita elinajanodotteesta tai heikkoudesta, joka perustuu tiukasti alkamisikään.
SMA on yleisin imeväiskuolleisuuden geneettinen syy.
mikä on SMA: ta koskevan tutkimuksen tilanne?
tutkimuksessa on keskitytty strategioihin, joilla pyritään lisäämään elimistön tuottamaa SMN-proteiinia, joka puuttuu taudin kromosomiin 5 liittyvistä muodoista. Lähestymistapoja ovat muun muassa menetelmät, joilla autetaan motoneuroneja selviytymään epäsuotuisissa olosuhteissa.
Joulukuuta. 23, 2016, Yhdysvaltain elintarvike-ja lääkevirasto (FDA) hyväksyi Spinrazan (nusinersen) SMA: n hoitoon. Spinraza on suunniteltu hoitamaan SMA: n taustalla olevaa vikaa, mikä tarkoittaa, että se saattaa olla tehokas hidastamaan, pysäyttämään tai ehkä kääntämään SMA: n oireet. Katso lisätietoja kohdasta Spinraza on hyväksytty.
toukokuussa 2019 FDA hyväksyi tsolgensman (onasemnogene abeparvovac-xioi), joka on ensimmäinen hermolihassairauden geenikorvaushoito. Tsolgensma on kertaluonteinen laskimonsisäinen infuusio alle 2-vuotiaille SMA: ta sairastaville lapsipotilaille, joilla on bi alleelisia mutaatioita SMN1-geenissä, mukaan lukien potilaat, jotka ovat diagnoosihetkellä presymptomaattisia. Lisätietoja: FDA hyväksyy Avexisin Tsolgensman spinaalisen lihasatrofian hoitoon lapsipotilailla.
lisätietoja, KS. Research, SMA: Full Speed Ahead and in Focus: Spinal Muscular Atrofy (SMA). SMA: n kanssa asuvien perheiden tarinoita löydät MDA: n blogista Strongly.
elokuussa 2020 FDA hyväksyi risdiplamin (tuotenimi Evrysdi*) SMA: n hoitoon aikuisilla ja kahden kuukauden ikäisillä tai sitä vanhemmilla lapsilla. Evysdi on suun kautta otettava lääke, joka on suunniteltu lisäämään SMN-proteiinin tasoja tehostamalla tuotantoa SMN2 ”backup” – geenistä.
- Ogino, S. & Wilson, R. B. Genetic testing and risk assessment for spinal muscular atrofy (SMA). Human Genetics (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Spinaalisen lihasatrofiaa määrittävän geenin tunnistaminen ja luonnehtiminen. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. non-5q spinal muscular atrophies: alfanumeerinen keitto sakenee. Neurology (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. Spinal muscular atrofy: Diagnosis and management in a new therapeutic era. Muscle and Nerve (2015). doi: 10.1002 / mus.24497