- Prélèvement d’échantillons, construction de bibliothèques et séquençage
- Contrôle de la qualité
- Estimation de la taille du génome
- Assemblage et évaluation du génome
- Annotation d’éléments répétitifs
- Annotation de gènes codant pour les protéines
- Annotation de fonction génique
- Construction de familles de gènes et reconstruction de la phylogénie
Prélèvement d’échantillons, construction de bibliothèques et séquençage
ADN génomique a été obtenu à partir d’un spécimen mâle de C. crocuta (NCBI taxonomy ID: 9678; Fig. 1) stocké dans le Zoo Frozen® du San Diego Zoo Institute for Conservation Research, États-Unis (ID du Zoo Frozen: KB4526).
L’ADN génomique a été extrait par phénol-chloroforme suivi d’une purification par précipitation à l’éthanol 13. L’ADN extrait a été exécuté et visualisé sur un 1.gel d’agarose à 5% exécuté dans un tampon 1x TBE pour vérifier la présence d’ADN de haut poids moléculaire. La concentration et la pureté de l’ADN ont été quantifiées sur un spectrophotomètre NanoDrop 2000 et un fluoromètre Qubit 2.0 (Thermo Fisher Scientific, États-Unis) avant d’être expédiées à BGI-Shenzhen, en Chine. Nous avons obtenu un total de 372 µg d’ADN génomique, avec une concentration de 0,418 µg / µL en utilisant le Nanodrop 2000 et de 0,245 à 0,399 µg / µL sur la base de quatre lectures répliquées à l’aide du fluoromètre Qubit 2.0. Le rapport de pureté 260/280 était de 1,95. Nous avons ensuite codé l’échantillon à l’aide du gène cytochrome b (Cytb). Ensuite, selon la stratégie de bibliothèque de gradients, nous avons construit 13 bibliothèques de taille d’insertion, avec les longueurs de taille d’insertion suivantes: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Nous avons utilisé le HiSeq. Séquenceur 2000 (Illumina, États-Unis) pour séquencer les lectures d’extrémité appariée (PE) pour chaque bibliothèque sur 14 voies. Un total d’environ 299 Go de données brutes a été généré à partir de 13 bibliothèques, atteignant une profondeur de séquençage (couverture) de 149,25 (tableau 1).
Contrôle de la qualité
Pour minimiser les erreurs de mauvais assemblage, nous avons filtré les lectures brutes avant l’assemblage du génome de novo selon les deux critères suivants. Tout d’abord, les lectures avec plus de 10 pb alignées sur la séquence de l’adaptateur (permettant < = 3 pb de non-concordance) ont été supprimées. Deuxièmement, les lectures avec 40% de bases ayant une valeur de qualité inférieure ou égale à 10 ont été rejetées. Enfin, nous avons obtenu des données de 190,4 G avec une couverture de 95,2 (tableau 2).
Estimation de la taille du génome
Trois bibliothèques à insertion courte (deux de 170 pb et une de 500 pb) ont été utilisées pour estimer la taille du génome et l’hétérozygotie à l’échelle du génome par analyse k-mer. Un total d’environ 385 M de lectures de PE a été soumis à jellyfish14 pour calculer la fréquence k-mer. Ensuite, la distribution k-mer a été illustrée par Genomescope7 avec des paramètres « k = 17; longueur = 100; couverture maximale = 1000 ». Nous avons obtenu une taille de génome estimée à 2 003 681 234 pb, et une hétérozygotie de 0,325% (Fig. 2).

17- estimation mer de la taille du génome. L’axe des abscisses est la profondeur (X), l’axe des ordonnées est la proportion qui représente la fréquence à cette profondeur divisée par la fréquence totale de toutes les profondeurs de couverture. Sans tenir compte du taux d’erreur de séquence, du taux d’hétérozygotie et du taux de répétition du génome, la distribution 17-mer devrait se rapprocher d’une distribution de Poisson.
Assemblage et évaluation du génome
SOAPdenovo (V1.06) 15 a été utilisé pour assembler le génome de novo, en suivant le filtrage des données de taille d’insertion courte et en supprimant le petit pic des données de taille d’insertion grande. L’algorithme d’assemblage de SOAPdenovo comprenait trois étapes principales. (1) Construction de Contig: les données de la bibliothèque de taille d’insertion courte ont été divisées en k-mers et construites à l’aide d’un graphique de Bruijn, ce qui a été simplifié en supprimant les pointes, en fusionnant les bulles, en supprimant la faible couverture de la connexion et en supprimant les petites répétitions. Nous avons obtenu la séquence de contig en connectant le trajet k-mer, ce qui donne un contig N50 de 2 104 pb et une longueur totale de 2 295 545 898 pb. (2) Construction d’échafaudages: nous avons obtenu 80% de toutes les lectures d’extrémité appariées alignées en réalignant toutes les lectures utilisables sur les contigs. Ensuite, nous avons calculé la quantité de relations d’extrémités appariées partagées entre chaque paire de contigs, pondéré le taux d’extrémités appariées cohérentes et conflictuelles, puis construit les échafaudages étape par étape. En conséquence, nous avons obtenu des échafaudages avec un N50 de 7 168 038 pb et une longueur totale de 2 355 303 269 pb à partir d’extrémités appariées de taille courte à des extrémités appariées de longue distance. (3) Fermeture des lacunes: Pour combler les lacunes à l’intérieur des échafaudages construits, nous avons utilisé les informations d’extrémité appariées pour récupérer les paires de lecture afin de refaire un assemblage local pour ces lectures collectées. En résumé, nous avons comblé 87,7 % des lacunes intra-échafaudages, soit 85,8 % de la longueur de l’écart de somme. La taille du contig N50 est passée de 2 104 pb à 21 301 pb (tableau 3). La taille de l’assemblage de l’échafaudage était de 2 355 303 269 pb, ce qui est proche de la taille du génome basée sur l’assemblage de 2 374 716 107 pb signalée pour la hyène rayée, Hyaena hyaena11 (accession NCBI: ASM300989v1). Nous avons également récupéré et annoté le génome mitochondrial de la hyène tachetée à l’aide du programme mitoz16, qui a une longueur de 16 858 pb, similaire aux premiers génomes mitochondriaux séquencés pour cette espèce12.
L’évaluation du projet de génome a été réalisée en examinant l’exhaustivité des orthologues à copie unique à l’aide de BUSCO (version 3.1.0) 17, en recherchant la base de données Mammaliaodb9 qui contient 4 104 groupes d’orthologues à copie unique. Au total, 95,5% des orthologues ont été identifiés comme complets, 2,5% comme fragmentés et 2,0% comme manquants, ce qui indique une qualité globale élevée de l’assemblage du génome de la hyène tachetée. Étant donné que 99,95% des échafaudages courts (< 1k) n’en abritaient que 1.2% de la longueur totale du génome, nous avons exclu ces échafaudages pour l’analyse en aval, y compris l’annotation d’éléments répétitifs et de caractéristiques génétiques.
Annotation d’éléments répétitifs
Les répétitions en tandem et les éléments transposables (TE) ont été recherchés et identifiés dans le génome de C. crocuta. Les répétitions en tandem ont été identifiées à l’aide du chercheur de répétitions en tandem (TRF, v4.07)18 et les éléments transposables (TES) ont été identifiés par une combinaison d’approches basées sur l’homologie et de novo. Pour la prédiction basée sur l’homologie, nous avons utilisé la version 4.0 de RepeatMasker.619 avec les paramètres « -nolow-no_is-norna-engine ncbi » et RepeatProteinMask (un programme dans le package RepeatMasker) avec les paramètres « -engine ncbi-noLowSimple-pvalue 0.0001 » pour rechercher des TES au niveau des nucléotides et des acides aminés sur la base de répétitions connues (Fig. 3). RepeatMasker a été utilisé pour l’identification au niveau de l’ADN à l’aide d’une bibliothèque personnalisée qui combinait l’ensemble de données Repbase21.1020. Au niveau des protéines, RepeatProteinMask a été utilisé pour effectuer RMBlast contre la base de données de protéines TE. Pour la prédiction ab initio, RepeatModeler(v1.0.8) 21 et LTR_FINDING(v1.06) 22 ont été appliqués pour construire la bibliothèque de répétition de novo. Les séquences de contamination et de copies multiples de la bibliothèque ont été supprimées et les séquences restantes ont été classées en fonction du résultat de l’EXPLOSION après alignement sur la base de données SwissProt. Sur la base de cette bibliothèque, nous avons utilisé RepeatMasker pour masquer les TES homologues et les avons classés (Fig. 4). Au total, un total de 826 Mb d’éléments répétitifs ont été identifiés chez la hyène tachetée, représentant 35,29% du génome entier (tableau 4).

Distribution du taux de divergence de chaque type d’élément transposable (TE) dans l’ensemble du génome de Crocuta crocuta basée sur une prédiction basée sur l’homologie. Le taux de divergence a été calculé entre les TES identifiés dans le génome à l’aide d’une méthode basée sur l’homologie et la séquence de consensus dans la base de données repbase20.

Distribution du taux de divergence de chaque type de TE dans l’ensemble du génome de Crocuta crocuta basée sur la prédiction ab initio. Le taux de divergence a été calculé entre les TE identifiées dans le génome par prédiction ab initio et la séquence de consensus dans la bibliothèque de TE prédite (voir Méthodes).
Annotation de gènes codant pour les protéines
Nous avons utilisé des approches de prédiction ab initio et basées sur des homologues pour annoter des gènes codant pour les protéines ainsi que des sites d’épissage et des isoformes d’épissage alternatives. La prédiction Ab initio a été réalisée sur le génome masqué à répétition à l’aide de modèles de gènes provenant d’humains, de chiens domestiques et de chats domestiques utilisant AUGUSTUS (version 2.5.5) 23, GENSCAN24, GlimmerHMM (version 3.0.4) 25 et SNAP (version 2006-07-28) 26, respectivement. Un total de 22 789 gènes ont été identifiés par cette méthode. Les protéines homologues d’Homo sapiens, de Felis catus et de Canis familiaris (de la version Ensembl 96) ont été cartographiées sur le génome de la hyène tachetée en utilisant tblastn (Blastall 2.2.26) 27 avec les paramètres « -e 1e-5 ». Les séquences alignées ainsi que leurs protéines d’interrogation ont ensuite été soumises à GeneWise (version 2.4.1) 28 pour rechercher un alignement épissé précis. L’ensemble de gènes final (22 747) a été collecté en fusionnant des résultats basés sur ab initio et des résultats basés sur des homologues à l’aide d’un pipeline personnalisé (tableau 5).
Annotation de fonction génique
Les fonctions géniques ont été attribuées selon la meilleure correspondance obtenue en alignant des séquences codantes de gènes traduites à l’aide de BLASTP avec les paramètres « -e 1e-5 » sur les bases de données SwissProt et Trembll (version Uniprot 2017-09). Les motifs et domaines des gènes ont été déterminés par Interposcan (v5)29 à partir de bases de données de protéines, notamment ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 et PROSITE35. Les ID d’ontologie des gènes pour chaque gène ont été obtenus à partir des entrées SwissProt et Trembll correspondantes. Tous les gènes étaient alignés sur les protéines KEGG, et la voie dans laquelle le gène pourrait être impliqué était dérivée des gènes appariés dans la base de données KEGG36. En résumé, 22 166 (97,45 %) des gènes codant les protéines prédits ont été annotés avec succès par au moins une des six bases de données (tableau 6).
Construction de familles de gènes et reconstruction de la phylogénie
Pour mieux comprendre l’histoire phylogénétique et l’évolution des familles de gènes de Crocuta crocuta, nous avons regroupé des séquences de gènes de sept espèces (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) et Homo sapiens en tant que groupe externe (Ensembl release-96, Panthera leo à partir de données non publiées) en familles de gènes en utilisant orthoMCL (v2.0.9) 37. Les gènes codant les protéines pour les huit espèces ont été récupérés en sélectionnant l’isoforme de transcription la plus longue pour chaque gène pour une affectation par paire en aval (construction de graphes). Nous avons effectué une recherche BLASTP tout contre tout sur les séquences protéiques de toutes les espèces de référence, avec un seuil de valeur E de 1e-5. La construction de familles de gènes a utilisé l’algorithme MCL38 avec le paramètre d’inflation de ‘1,5’. Au total, 16 271 familles de gènes de C. crocuta, H. sapiens, F. catus, A. melanoleuca ont été regroupées. Il y avait 11 671 familles de gènes partagées par ces quatre espèces, tandis que 292 familles de gènes contenant 1 446 gènes étaient spécifiques de C. Crocuta (Fig. 5). De manière notable, les familles de gènes partagées par C. crocuta et F. catus étaient inférieures à celles partagées par C. crocuta et H. sapiens, ce qui pourrait résulter du fait que H. sapiens avait un génome et une annotation plus complets.
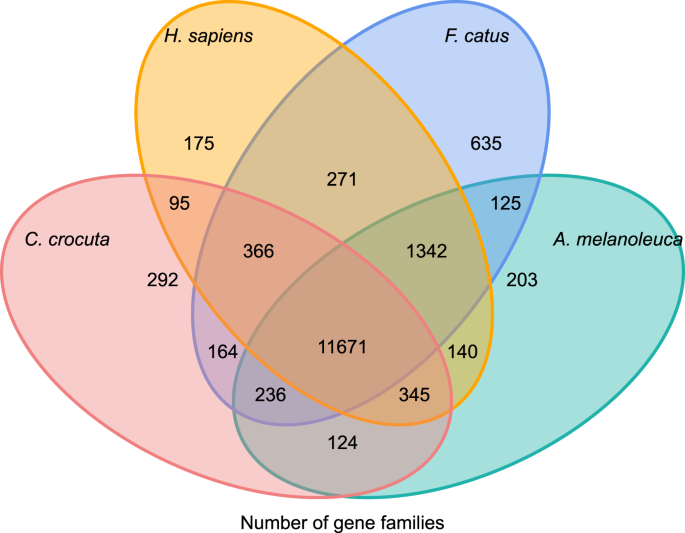
Diagramme de Venn montrant la comparaison de gènes codant des protéines partagés et uniques chez la hyène tachetée, l’homme, le chat domestique et le chien domestique sur la base d’une analyse orthologique.
Nous avons identifié 6 601 gènes orthologues à copie unique pour reconstruire l’arbre phylogénétique des huit espèces. Des alignements de séquences multiples de séquences d’acides aminés pour chaque gène ont été générés à l’aide du MUSCLE (version 3.8.31) 39, et rognés à l’aide de blocs G (0,91b) 40, obtenant des régions bien alignées avec les paramètres « -t = p-b3 = 8-b4 = 10-b5 = n-e = -st ». Nous avons effectué une analyse phylogénétique en utilisant la méthode du maximum de vraisemblance mise en œuvre dans PhyML (v3.0)41, en utilisant le modèle JTT + G +I pour la substitution d’acides aminés (Fig. 6). La racine de l’arbre a été déterminée en minimisant la hauteur de l’arbre entier via Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Enfin, nous avons estimé le temps de divergence entre les huit lignées en utilisant MCMCTree du logiciel PAML version 4.442. Deux priors basés sur les archives fossiles ont été utilisés pour calibrer le taux de substitution, y compris Boreoeutheria (91-102 MYA) et Carnivora (52-57 MYA) 43. Conformément aux études précédentes, les groupes de hyènes tachetées avec les quatre espèces incluses des Félidés dans un clade définissant le sous-ordre des Feliformia, qui a divergé des Caniformia (représentés par le chien domestique et le panda géant) 53.9 Mya44.

Arbre phylogénétique de C. crocuta et de sept autres espèces construit selon la méthode du maximum de vraisemblance basée sur 6 601 orthologues à copie unique. Le temps de divergence a été estimé à l’aide des deux prieurs d’étalonnage dérivés de la base de données de l’arbre temporel (http://www.timetree.org), qui sont marqués par un losange rouge. Tous les temps de divergence estimés sont indiqués avec des intervalles de confiance de 95% entre parenthèses.