Atrofia muscolare spinale
Scarica il nostro foglio informativo Atrofia muscolare spinale
Scopri la risposta COVID-19 di MDA
Che cos’è l’atrofia muscolare spinale?
L’atrofia muscolare spinale (SMA) è una malattia genetica che colpisce il sistema nervoso centrale, il sistema nervoso periferico e il movimento muscolare volontario (muscolo scheletrico).
La maggior parte delle cellule nervose che controllano i muscoli si trovano nel midollo spinale, che rappresenta la parola spinale nel nome della malattia. SMA è muscolare perché il suo effetto primario è sui muscoli, che non ricevono segnali da queste cellule nervose. Atrofia è il termine medico per ottenere più piccolo, che è ciò che generalmente accade ai muscoli quando non sono stimolati dalle cellule nervose.
La SMA comporta la perdita di cellule nervose chiamate motoneuroni nel midollo spinale ed è classificata come malattia del motoneurone.
Nella forma più comune di SMA (SMA cromosoma 5 o SMA correlata a SMN), vi è un’ampia variabilità nell’età di insorgenza, nei sintomi e nella velocità di progressione. Per tenere conto di queste differenze, la SMA correlata al cromosoma 5, che spesso è autosomica recessiva, è classificata nei tipi da 1 a 4.
L’età in cui iniziano i sintomi della SMA è approssimativamente correlata al grado in cui la funzione motoria è influenzata: prima è l’età di insorgenza, maggiore è l’impatto sulla funzione motoria. I bambini che mostrano sintomi alla nascita o nell’infanzia hanno in genere il livello più basso di funzionamento (tipo 1). La SMA ad esordio tardivo con un decorso meno grave (tipi 2 e 3, e negli adolescenti o negli adulti, tipo 4) è generalmente correlata a livelli sempre più elevati di funzione motoria.
Per ulteriori informazioni, vedere Forme di SMA.
Quali sono le cause SMA?
Il cromosoma 5 SMA è causato da una carenza di una proteina del motoneurone chiamata SMN, per “sopravvivenza del motoneurone.”Questa proteina, come suggerisce il nome, sembra essere necessaria per la normale funzione del motoneurone. SMN svolge un ruolo fondamentale nell’espressione genica nei motoneuroni. La sua carenza è causata da difetti genetici (mutazioni) sul cromosoma 5 in un gene chiamato SMN1. La mutazione più comune nel gene SMN1 all’interno dei pazienti con diagnosi di SMA è una delezione di un intero segmento, chiamato esone 7.1 I geni SMN2 vicini possono in parte compensare i geni SMN1 non funzionali in quanto vi è un’identità del 99% tra questi due geni.2
Altre forme rare di SMA (non cromosomica 5) sono causate da mutazioni in geni diversi da SMN1.3
Per ulteriori informazioni, inclusa una lettura dettagliata sulla SMA rara, non legata al cromosoma 5, vedere Forme di SMA e cause/ereditarietà.
Quali sono i sintomi della SMA?
I sintomi SMA coprono un ampio spettro, che va da lieve a grave.
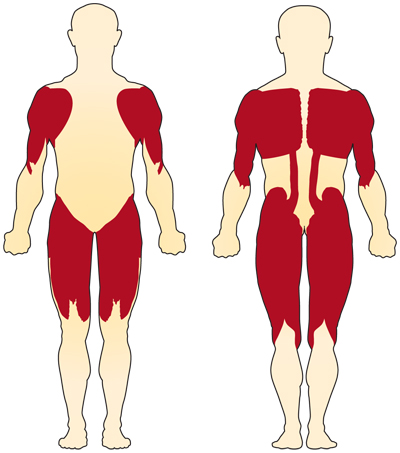
Il sintomo principale della SMA correlata al cromosoma 5 (SMN-related) è la debolezza dei muscoli volontari. I muscoli più colpiti sono quelli più vicini al centro del corpo, come quelli delle spalle, fianchi, cosce e parte superiore della schiena. Gli arti inferiori sembrano essere colpiti più degli arti superiori e i riflessi tendinei profondi sono diminuiti.4
Complicazioni speciali si verificano se i muscoli utilizzati per la respirazione e la deglutizione sono interessati, con conseguenti anomalie in queste funzioni. Se i muscoli della schiena si indeboliscono, possono svilupparsi curvature spinali.
C’è una grande quantità di variazione nell’età di insorgenza e nel livello di funzione motoria raggiunto nella SMA correlata al cromosoma 5. Questi sono approssimativamente correlati con la quantità di proteina SMN funzionale presente nei motoneuroni, che a sua volta è correlata con quante copie di geni SMN2 una persona ha. Il funzionamento sensoriale, mentale ed emotivo è del tutto normale nel cromosoma-5 SMA.
Alcune forme di SMA non sono legate al cromosoma 5 o alla carenza di SMN. Queste forme variano notevolmente in gravità e nei muscoli più colpiti. Mentre la maggior parte delle forme, come la forma correlata al cromosoma 5, colpisce principalmente i muscoli prossimali, esistono altre forme che colpiscono principalmente i muscoli distali (quelli più lontani dal centro del corpo)-almeno all’inizio.
Per ulteriori informazioni, vedere Segni e sintomi.
Qual è la progressione della SMA?
Nella SMA correlata al cromosoma 5, più tardi iniziano i sintomi e più proteina SMN c’è, più lieve è il decorso della malattia. Mentre in passato, i bambini con SMA in genere non sopravvivevano più di due anni, oggi la maggior parte dei medici considera la SMA correlata alla SMN come un continuum e preferisce non fare previsioni rigide sull’aspettativa di vita o sulla debolezza basate rigorosamente sull’età di insorgenza.
La SMA è la causa genetica più comune di mortalità nei neonati.
Qual è lo stato della ricerca sulla SMA?
La ricerca si è concentrata su strategie per aumentare la produzione del corpo della proteina SMN priva delle forme correlate al cromosoma 5 della malattia. Gli approcci includono metodi per aiutare i motoneuroni a sopravvivere in circostanze avverse.
Il dic. 23, 2016, la Food and Drug Administration (FDA) degli Stati Uniti ha approvato Spinraza (nusinersen) per il trattamento della SMA. Spinraza è progettato per trattare il difetto sottostante nella SMA, il che significa che potenzialmente può essere efficace nel rallentare, arrestare o forse invertire i sintomi della SMA. Per ulteriori informazioni, vedere Spinraza è approvato.
A maggio 2019, la FDA ha approvato Zolgensma (onasemnogene abeparvovac-xioi), la prima terapia genica sostitutiva per una malattia neuromuscolare. Zolgensma è un’infusione endovenosa una tantum (nella vena) per il trattamento di pazienti pediatrici di età inferiore ai 2 anni con SMA con mutazioni bi-alleliche nel gene SMN1, compresi quelli che sono presintomatici alla diagnosi. Per ulteriori informazioni, leggi FDA approva lo Zolgensma di AveXis per il trattamento dell’atrofia muscolare spinale nei pazienti pediatrici.
Per ulteriori informazioni, vedere Research, SMA: Full Speed Ahead and In Focus: Spinale Atrofia muscolare (SMA). Per le storie di famiglie che vivono con SMA, vedere le nostre storie SMA su Strongly, il blog MDA.
Nell’agosto 2020, la FDA ha approvato risdiplam (marca Evrysdi*) per il trattamento della SMA negli adulti e nei bambini di due mesi di età o più. Evysdi è un farmaco orale progettato per aumentare i livelli della proteina SMN migliorando la produzione dal gene “backup” SMN2.
- Ogino, S. & Wilson, R. B. Test genetici e valutazione del rischio per atrofia muscolare spinale (SMA). Genetica umana (2002). doi:10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identificazione e caratterizzazione di un gene che determina l’atrofia muscolare spinale. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. atrofie muscolari spinali non 5q: la zuppa alfanumerica si addensa. Neurologia (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. Atrofia muscolare spinale: diagnosi e gestione in una nuova era terapeutica. Muscoli e nervi (2015). doi:10.1002 / mus.24497