Sample collection, library construction and sequencing
Genomic DNA was obtained from a male specimen of C. crocuta(NCBI taxonomy ID: 9678; Fig. 1) armazenado no zoológico congelado® no San Diego Zoo Institute for Conservation Research, EUA (ID do Zoo congelado: KB4526).
o ADN genómico foi extraído utilizando fenol-clorofórmio seguido de purificação utilizando precipitação de etanol 13. O ADN extraído foi executado e visualizado num 1.O gel de agarose a 5% é aplicado em 1x tampão TBE para verificar a presença de ADN de elevada massa molecular. A concentração e pureza do ADN foram quantificadas num espectrofotómetro de nanodrop 2000 e num Fluorómetro de Qubit 2.0 (Thermo Fisher Scientific, EUA) antes de serem enviados para BGI-Shenzhen, China. Obteve-se um total de 372 µg de DNA genômico, com uma concentração de 0.418 µg/µL, utilizando o Nanodrop 2000 e 0.245–0.399 µg/µL, com base em quatro replicar leituras utilizando o Qubit 2.0 Fluorímetro. O rácio de pureza de 260/280 era de 1,95. Em seguida, marcamos a amostra usando o gene citocromo b (Cytb). Então, de acordo com a estratégia da biblioteca gradiente, construímos 13 bibliotecas de tamanho de inserção, com os seguintes comprimentos de tamanho de inserção: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Usámos o HiSeq. 2000 sequencer (Illumina, EUA) to sequence Emparelhed-End (PE) lê para cada biblioteca em 14 faixas. Um total de cerca de 299 GB de dados brutos foi gerado a partir de 13 bibliotecas, alcançando uma profundidade de sequenciamento (cobertura) de 149,25 (Tabela 1).
Tabela 1 estatísticas de dados de leitura bruta, assumindo que o tamanho do genoma é de 2.0 Gb.
controle de qualidade
para minimizar erros de montagem, filtramos leituras em bruto antes da montagem do genoma de novo de acordo com os dois critérios seguintes. Em primeiro lugar, leituras com mais de 10 bp alinhadas à sequência do adaptador (permitindo <= 3 BP desfasamento) foram removidas. Em segundo lugar, lê-se que 40% das bases com um valor de qualidade inferior ou igual a 10 foram descartadas. Por último, obtivemos dados de 190,4 G com uma cobertura de 95,2 (Quadro 2).
Quadro 2 estatísticas de dados na sequência da filtragem dos dados de leitura bruta.
Estimation of genome size
Three short-insert libraries (two of 170 bp and one of 500 bp) were used to estimate the genome size and genome-wide heterozygosity by k-mer analysis. Um total de cerca de 385 m de leitura PE foram submetidos a jellyfish14 para calcular a frequência K-mer. Em seguida, a distribuição K-mer foi ilustrada por Genomescope7 com parâmetros “k = 17; Comprimento = 100; Cobertura máxima = 1000”. Nós obtivemos um tamanho estimado de genoma 2.003.681.234 bp ,e heterozigosidade de 0,325% (Fig. 2).
Fig. 2
17-uma estimativa do tamanho do genoma. O eixo x é a profundidade (X), o eixo y é a proporção que representa a frequência a essa profundidade dividida pela frequência total de todas as profundidades de cobertura. Sem considerar a taxa de erro de sequência, a taxa de heterozigosidade e a taxa de repetição do genoma, a distribuição de 17-mer deve aproximar-se de uma distribuição de Poisson.
genoma assembly and assessment
SOAPdenovo (V1.06) 15 foi empregado para montar o genoma de novo, após a filtragem dos dados de tamanho de inserção curto e removendo o pequeno pico dos dados de tamanho de inserção grande. O algoritmo de montagem de SOAPdenovo incluiu três etapas principais. (1) Contig construction: the short-insert size library data were split into k-mers and constructed using a De Bruijn graph, which was simplified by removing tips, merging bubbles, removing the low coverage of the connection and removing small repeats. Obtivemos a sequência contig ligando o caminho k-mer, resultando em um contig N50 2,104 bp, e comprimento total 2,295,545,898 bp. (2) construção de andaimes: obtivemos 80% de todas as leituras alinhadas emparelhadas realinhando todas as leituras utilizáveis em contigs. Em seguida, calculamos a quantidade de relacionamentos compartilhados emparelhados entre cada par de contigs, ponderou a taxa de pontas emparelhadas consistentes e conflitantes, e, em seguida, construiu os andaimes passo a passo. Como resultado, obtivemos andaimes com N50 7,168,038 bp, e comprimento total 2,355,303,269 bp de extremidades emparelhadas curtas de tamanho de inserção, para extremidades emparelhadas longas e distantes. (3) fechamento de lacunas: para preencher as lacunas dentro dos andaimes construídos, usamos a informação emparelhada para recuperar os pares de leitura para fazer uma montagem local novamente para estas leituras coletadas. Em resumo, fechamos 87,7% das aberturas intra-andaimes, ou 85,8% do comprimento total das aberturas. A dimensão do contig N50 aumentou de 2,104 bp para 21,301 bp (Quadro 3). O tamanho da montagem do andaime foi de 2,355,303,269 bp, que é próximo ao tamanho do genoma baseado na montagem de 2,374,716,107 bp relatado para a hiena listrada, Hiaena hyaena11 (adesão NCBI: ASM300989v1). Nós também recuperamos e anotamos o genoma mitocondrial da hiena manchada usando o programa Mitoz16, que tem um comprimento de 16.858 bp, similar aos primeiros genomas mitocondriais sequenciados para esta especia12.
Quadro 3 estatísticas do comprimento da sequência montada.
a avaliação do rascunho do genoma foi realizada olhando para a completude de ortólogos de cópia única usando o BUSCO (Versão 3.1.0)17, pesquisando a base de dados Mammaliaodb9, que contém 4.104 grupos ortolog de cópia única. Um total de 95,5% dos ortólogos foram identificados como completos, 2,5% como fragmentados e 2,0% como desaparecidos, indicando uma alta qualidade global da montagem do genoma da hiena manchada. Dado que 99,95% dos andaimes curtos (<1k) abrigavam apenas 1.2% do comprimento total do genoma, excluímos estes andaimes para análise a jusante, incluindo a anotação de elementos repetitivos e características genéticas.
anotação de elementos repetitivos
tanto repetições tandem como elementos transposíveis (TE) foram pesquisados e identificados através do genoma de C. crocuta. As repetições Tandem foram identificadas usando o localizador de repetições Tandem (TRF, v4.07)18 e elementos transponíveis (ses) foram identificados por uma combinação de abordagens baseadas em homologia e de novo. Para a previsão baseada em homologia, usamos a versão 4.0 do RepeatMasker.619 with the settings “- nolow-no_is-norna-engine ncbi” and RepeatProteinMask (a program within RepeatMasker package) with the settings “- engine ncbi-noLowSimple-pvalue 0.0001” to search TEs at the nucleotide and amino acid level based on known repeats (Fig. 3). Repetmasker foi aplicado para identificação de nível de DNA usando uma biblioteca personalizada que combinou o conjunto de dados Repbase21.1020. No nível de proteína, Repetproteinmask foi usado para executar RMBlast contra a base de dados de proteína TE. Para a previsão ab initio, Repetmodeler (v1.0.8)21 e LTR_FINDING (v1.06) 22 foram aplicados para a construção da biblioteca de novo repeat. A contaminação e sequências multi-cópias na biblioteca foram removidas e as sequências restantes foram classificadas de acordo com o resultado da explosão após o alinhamento à base de dados Swisssprot. Com base nesta biblioteca, usamos o Repetmasker para mascarar os TEs homólogos e classificá-los (Fig. 4). Globalmente, um total de 826 Mb de elementos repetitivos foram identificados na hiena manchada, compreendendo 35,29% de todo o genoma (Quadro 4).
Fig. 3
distribuição da taxa de divergência de cada tipo de elemento transponível (TE) no conjunto do genoma Crocuta crocuta baseado na previsão homológica. A taxa de divergência foi calculada entre as e-a identificadas no genoma utilizando um método baseado em homologia e a sequência de consenso na base de dados Repbase 20.
Fig. 4
distribuição da taxa de divergência de cada tipo de TE na montagem do genoma Crocuta crocuta com base na previsão ab initio. A taxa de divergência foi calculada entre as e-a identificadas no genoma pela previsão ab initio e a sequência de consenso na biblioteca te prevista (ver Métodos).
Quadro 4 teor de elementos transponíveis da montagem do genoma Crotuta crotuta.
foi utilizada a predição ab initio e abordagens homolog-based para anotar genes codificadores de proteínas, bem como locais de splicing e isoformas de splicing alternativas. A predição Ab initio foi realizada no genoma de repetição Mascarado usando modelos genéticos de humanos, cães domésticos e gatos domésticos usando AUGUSTUS (versão 2.5.5)23, GENSCAN24, GlimmerHMM (versão 3.0.4)25, e SNAP (versão 2006-07-28)26, respectivamente. Um total de 22.789 genes foram identificados por este método. Proteínas homólogas de, Homo sapiens, Felis catus e Canis familiaris (a partir da liberação de Ensembl 96) foram mapeadas para o genoma da hiena manchada usando tblastn (Blastall 2.2.26)27 com parâmetros “-e 1e-5”. As sequências alinhadas, bem como suas proteínas de consulta foram então submetidas a trechos (versão 2.4.1)28 para pesquisar um alinhamento esplicado preciso. O conjunto final de genes (22.747) foi coletado pela fusão dos resultados ab initio e homolog baseados em um pipeline personalizado (Tabela 5).
Quadro 5 estatísticas gerais do número de genes codificadores de proteínas com base em ab initio (de novo) e métodos de previsão baseados em homologia.
Gene função de anotação
Gene funções foram atribuídas de acordo com o melhor resultado obtido pelo alinhamento traduzido gene de codificação de sequências usando BLASTP com os parâmetros “-e 1e-5” para o SwissProt e TrEMBL bancos de dados (Uniprot lançamento 2017-09). Os motivos e domínios dos genes foram determinados por InterProScan (v5)29 contra bases de dados de proteínas, incluindo ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 e PROSITE35. Identificações ontológicas de genes para cada gene foram obtidas a partir das correspondentes entradas de Swisssprot e TrEMBL. Todos os genes foram alinhados com as proteínas KEGG, e a via na qual o gene pode estar envolvido foi derivada dos genes correspondentes na base de dados KEGG36. Em resumo, 22.166 (97.45%) dos genes codificadores de proteínas previstos foram anotados com sucesso por pelo menos uma das seis bases de Dados (Tabela 6).
Tabela 6 Número de genes com homologia prevista ou classificação funcional de acordo com o alinhamento a diferentes bases de dados de proteínas.
família do Gene da construção e reconstrução da filogenia
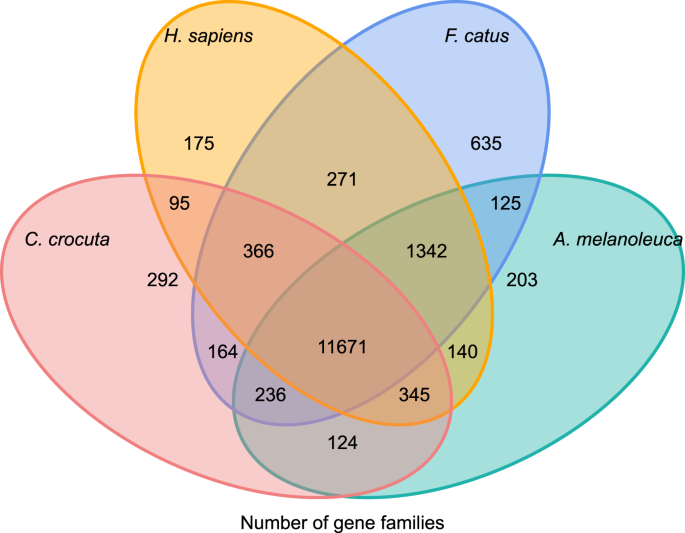
Para obter insights sobre o filogenética história e a evolução das famílias de genes de Crocuta crocuta, nós de cluster sequências de genes de sete espécies (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) e o Homo sapiens como o outgroup (Ensembl lançamento-96, Panthera leo, dos dados não publicados) em famílias de genes usando orthoMCL (v2.0.9)37. The protein-coding genes for the eight species were retrieved by selecting the longest transcript isoorm for each gene for downstream pairwise assignment (graph building). Realizamos uma pesquisa all-against-all BLASTP sobre as sequências proteicas de todas as espécies de referência, com um cut-off de valor E de 1e-5. A construção da família de genes empregou o algoritmo MCL 38 com o parâmetro de inflação de “1.5”. Um total de 16.271 famílias de genes de C. crocuta, H. sapiens, F. catus, A. melanoleuca foram agrupadas. Havia 11.671 famílias de genes compartilhadas por estas quatro espécies, enquanto 292 famílias de genes contendo 1.446 genes eram específicas de C. Crocuta(Fig. 5). Notavelmente, as famílias de genes C. crocuta e F. catus compartilhadas eram menos do que C. crocuta e H. sapiens compartilhadas, o que poderia resultar de que H. sapiens tinha um genoma mais completo e anotação.
Fig. 5
Diagrama de Venn mostrando a comparação de genes compartilhados e únicos codificadores de proteínas entre hiena manchada, humano, gato doméstico e cão doméstico com base na análise ortológica.
identificámos 6.601 genes ortólogos de cópia única para reconstruir a árvore filogenética das oito espécies. Múltiplos alinhamentos de sequências de aminoácidos para cada gene foram gerados usando músculo (versão 3.8.31)39, e aparados usando bloqueios (0.91 b)40, alcançando regiões bem alinhadas com os parâmetros “-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”. Nós realizamos a análise filogenética usando o método de máxima probabilidade implementado no PhyML (v3.0)41, usando o modelo JTT + g + I para substituição de aminoácidos(Fig. 6). A raiz da árvore foi determinada minimizando a altura de toda a árvore através do Treebest (v1)..9.2; http://treesoft.sourceforge.net/treebest.shtml). Finalmente, estimamos o tempo de divergência entre as oito linhagens usando MCMCTree da versão PAML 4.4 software package42. Dois priores baseados no registro fóssil foram usados para calibrar a taxa de substituição, incluindo Boreoeutheria (91-102 MYA) e Carnivora (52-57 MYA)43. Consistente com estudos anteriores, os grupos de hienas malhadas com as quatro espécies incluídas dos Felidae em um clado que define a subordem Feliformia, que divergiu da Caniformia (representada pelo cão doméstico e panda gigante) 53.9 Mya44.
Fig. 6
Phylogenetic tree of C. crocuta and seven other species constructed by the maximum likelihood method based on 6.601 single-copy orthologues. O tempo de divergência foi estimado usando os dois priores de calibração derivados da Base de dados da árvore do tempo (http://www.timetree.org), que são marcados por um losango vermelho. Todos os tempos de divergência estimados são mostrados com intervalos de confiança de 95% entre parêntesis.