Atrofia Muscular Espinal
Descargue nuestra Hoja Informativa sobre la Atrofia Muscular Espinal
Conozca la respuesta de MDA a la COVID-19
¿Qué es la atrofia muscular espinal?
La atrofia muscular espinal (AME) es una enfermedad genética que afecta al sistema nervioso central, al sistema nervioso periférico y al movimiento muscular voluntario (músculo esquelético).
La mayoría de las células nerviosas que controlan los músculos se encuentran en la médula espinal, lo que explica la palabra espinal en el nombre de la enfermedad. La AME es muscular porque su efecto principal es en los músculos, que no reciben señales de estas células nerviosas. Atrofia es el término médico para disminuir de tamaño, que es lo que generalmente le sucede a los músculos cuando no son estimulados por las células nerviosas.
La AME implica la pérdida de células nerviosas llamadas neuronas motoras en la médula espinal y se clasifica como una enfermedad de la neurona motora.
En la forma más común de AME (AME cromosómica 5, o AME relacionada con la AME), existe una amplia variabilidad en la edad de inicio, los síntomas y la tasa de progresión. Para tener en cuenta estas diferencias, la AME relacionada con el cromosoma 5, que a menudo es autosómica recesiva, se clasifica en los tipos 1 a 4.
La edad a la que comienzan los síntomas de AME se correlaciona aproximadamente con el grado en que la función motora se ve afectada: Cuanto más temprana sea la edad de inicio, mayor será el impacto en la función motora. Los niños que presentan síntomas al nacer o en la infancia suelen tener el nivel más bajo de funcionamiento (tipo 1). La AME de inicio tardío con un curso menos grave (tipos 2 y 3, y en adolescentes o adultos, tipo 4) generalmente se correlaciona con niveles cada vez más altos de función motora.
Para obtener más información, consulte Formas de SMA.
¿Qué causa la AME?
La SMA del cromosoma 5 es causada por una deficiencia de una proteína de la neurona motora llamada SMN, para «supervivencia de la neurona motora».»Esta proteína, como su nombre lo indica, parece ser necesaria para la función normal de la neurona motora. La NSM desempeña un papel fundamental en la expresión génica en las neuronas motoras. Su deficiencia es causada por defectos genéticos (mutaciones) en el cromosoma 5 en un gen llamado SMN1. La mutación más común en el gen SMN1 dentro de los pacientes diagnosticados con AME es una deleción de un segmento completo, llamado exón 7.1 Los genes SMN2 vecinos pueden compensar en parte los genes SMN1 no funcionales, ya que hay un 99% de identidad entre estos dos genes.2
Otras formas raras de AME (sin cromosoma 5) son causadas por mutaciones en genes distintos de la AME1.3
Para obtener más información, incluida una lectura detallada de AME poco frecuente sin cromosoma 5, consulte Formas de AME y Causas/Herencia.
¿Cuáles son los síntomas de la AME?
Los síntomas de AME abarcan un amplio espectro, que va de leves a graves.
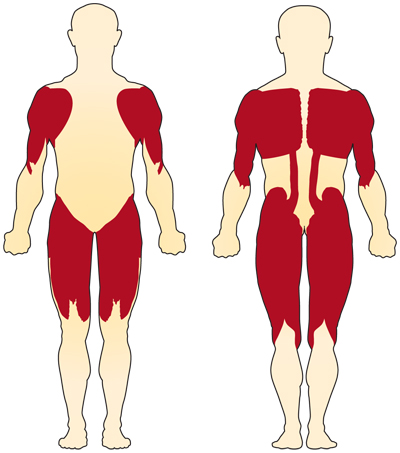
El síntoma principal de la AME relacionada con el cromosoma 5 (relacionada con la NSM) es la debilidad de los músculos voluntarios. Los músculos más afectados son los más cercanos al centro del cuerpo, como los de los hombros, las caderas, los muslos y la parte superior de la espalda. Los miembros inferiores parecen estar más afectados que los miembros superiores, y los reflejos tendinosos profundos disminuyen.4
Se producen complicaciones especiales si los músculos utilizados para respirar y tragar se ven afectados, lo que resulta en anomalías en estas funciones. Si los músculos de la espalda se debilitan, se pueden desarrollar curvaturas espinales.
Hay una gran variación en la edad de inicio y el nivel de función motora alcanzados en la AME relacionada con el cromosoma 5. Estos están aproximadamente correlacionados con cuánta proteína SMN funcional está presente en las neuronas motoras, lo que a su vez se correlaciona con cuántas copias de genes SMN2 tiene una persona. El funcionamiento sensorial, mental y emocional es completamente normal en la AME del cromosoma 5.
Algunas formas de AME no están vinculadas a la deficiencia del cromosoma 5 o de la NSM. Estas formas varían mucho en severidad y en los músculos más afectados. Si bien la mayoría de las formas, como la forma relacionada con el cromosoma 5, afectan principalmente a los músculos proximales, existen otras formas que afectan principalmente a los músculos distales (aquellos más alejados del centro del cuerpo), al menos al principio.
Para obtener más información, consulte Signos y síntomas.
¿Cuál es la progresión de la AME?
En la AME relacionada con el cromosoma 5, cuanto más tarde comiencen los síntomas y cuanta más proteína SMN haya, más leve será el curso de la enfermedad. Mientras que en el pasado, los bebés con AME normalmente no sobrevivían más de dos años, hoy en día la mayoría de los médicos consideran que la AME relacionada con la AME es un continuo y prefieren no hacer predicciones rígidas sobre la esperanza de vida o la debilidad basadas estrictamente en la edad de inicio.
La AME es la causa genética más común de mortalidad en lactantes.
¿Cuál es el estado de la investigación sobre AME?
La investigación se ha centrado en estrategias para aumentar la producción corporal de la proteína SMN que falta en las formas de la enfermedad relacionadas con el cromosoma 5. Los enfoques incluyen métodos para ayudar a las neuronas motoras a sobrevivir en circunstancias adversas.
En Diciembre. El 23 de septiembre de 2016, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó Spinraza (nusinersen) para el tratamiento de la AME. Spinraza está diseñado para tratar el defecto subyacente en la AME, lo que significa que puede ser eficaz para ralentizar, detener o tal vez revertir los síntomas de la AME. Para obtener más información, consulte Spinraza está aprobado.
En mayo de 2019, la FDA aprobó Zolgensma (onasemnogén abeparvovac-xioi), la primera terapia de reemplazo genético para una enfermedad neuromuscular. Zolgensma es una infusión intravenosa única (en la vena) para el tratamiento de pacientes pediátricos menores de 2 años de edad con AME con mutaciones bialélicas en el gen SMN1, incluso aquellos que son presintomáticos en el momento del diagnóstico. Para obtener más información, lea la FDA Aprueba Zolgensma de AveXis para el Tratamiento de la Atrofia Muscular Espinal en Pacientes Pediátricos.
Para obtener más información, consulte Investigación, SMA: Avance a toda velocidad y Enfoque: Atrofia Muscular Espinal (SMA). Para historias de familias que viven con AME, vea nuestras historias de AME en Strongly, el blog de MDA.
En agosto de 2020, la FDA aprobó risdiplam (nombre de marca Evrysdi*) para el tratamiento de la AME en adultos y niños de dos meses de edad o más. Evysdi es un medicamento oral diseñado para aumentar los niveles de la proteína SMN al mejorar la producción del gen «de respaldo» SMN2.
- Ogino, S. & Wilson, R. B. Pruebas genéticas y evaluación del riesgo de atrofia muscular espinal (AME). Human Genetics (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identificación y caracterización de un gen determinante de la atrofia muscular espinal. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. Atrofias musculares espinales no 5q: La sopa alfanumérica espesa. Neurología (2011). doi:10.1212 / WNL.0b013e3182267bd8
- Arnold, W. D., Kassar, D. & Kissel, J. T. Atrofia muscular espinal: Diagnóstico y manejo en una nueva era terapéutica. Muscle and Nerve (2015). doi: 10.1002 / mus.24497