- la raccolta del Campione, di costruzione di biblioteche e di sequenziamento
- Controllo di qualità
- Stima della dimensione del genoma
- Assemblaggio del genoma e la valutazione
- Annotazione degli elementi ripetitivi
- Annotazione genica codificante proteine
- Annotazione della funzione genica
- famiglia del Gene costruzione e la ricostruzione della filogenesi
la raccolta del Campione, di costruzione di biblioteche e di sequenziamento
il DNA Genomico è stato ottenuto da un esemplare maschio di C. crocuta (NCBI tassonomia ID: 9678; Fig. 1) conservato nel Frozen Zoo® presso il San Diego Zoo Institute for Conservation Research, USA (ID Zoo congelato: KB4526).
Il DNA genomico è stato estratto utilizzando fenolo-cloroformio seguito da purificazione mediante precipitazione di etanolo 13. Il DNA estratto è stato eseguito e visualizzato su un 1.il gel di agarosio al 5% viene eseguito in tampone 1x TBE per verificare la presenza di DNA ad alto peso molecolare. La concentrazione e la purezza del DNA sono state quantificate su uno spettrofotometro NanoDrop 2000 e un fluorometro Qubit 2.0 (Thermo Fisher Scientific, USA) prima della spedizione a BGI-Shenzhen, Cina. Abbiamo ottenuto un totale di 372 µg di DNA genomico, con una concentrazione di 0,418 µg/µL utilizzando il Nanodrop 2000 e 0,245-0,399 µg/µL sulla base di quattro letture replicate utilizzando il fluorometro Qubit 2.0. Il rapporto di purezza 260/280 era 1,95. Abbiamo quindi codificato il campione utilizzando il gene del citocromo b (Cytb). Quindi, secondo la strategia della libreria gradiente, abbiamo costruito 13 librerie di dimensioni insert, con le seguenti lunghezze di dimensioni insert: 170 bp, 500 bp, 800 bp, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Abbiamo usato il HiSeq. 2000 sequencer (Illumina, USA) per sequence Paired-End (PE) legge per ogni libreria attraverso 14 corsie. Un totale di circa 299 Gb di dati grezzi è stato generato da 13 librerie, raggiungendo una profondità di sequenziamento (copertura) di 149,25 (Tabella 1).
Controllo di qualità
Per ridurre al minimo gli errori di assemblaggio errato, abbiamo filtrato le letture grezze prima dell’assemblaggio del genoma de novo secondo i seguenti due criteri. Innanzitutto, sono state rimosse le letture con più di 10 bp allineate alla sequenza dell’adattatore (consentendo <= 3 bp non corrispondenti). In secondo luogo, le letture con 40% di basi con un valore di qualità inferiore o uguale a 10 sono state scartate. Infine, abbiamo ottenuto i dati 190.4 G con una copertura di 95.2 (Tabella 2).
Stima della dimensione del genoma
Tre librerie a inserto corto (due di 170 bp e una di 500 bp) sono state utilizzate per stimare la dimensione del genoma e l’eterozigosità a livello del genoma mediante analisi k-mer. Un totale di circa 385 M PE legge sono stati sottoposti a jellyfish14 per calcolare la frequenza k-mer. Quindi la distribuzione k-mer è stata illustrata da Genomescope7 con i parametri “k = 17; length = 100; max coverage = 1000”. Abbiamo ottenuto una dimensione stimata del genoma 2,003,681,234 bp, ed eterozigosità di 0,325% (Fig. 2).

17-mer stima della dimensione del genoma. L’asse x è la profondità (X), l’asse y è la proporzione che rappresenta la frequenza a quella profondità divisa per la frequenza totale di tutte le profondità di copertura. Senza considerare il tasso di errore di sequenza, il tasso di eterozigosità e il tasso di ripetizione del genoma, la distribuzione 17-mer dovrebbe approssimare una distribuzione di Poisson.
Assemblaggio del genoma e la valutazione
SOAPdenovo (V1.06) 15 è stato impiegato per assemblare il genoma de novo, dopo il filtraggio dei dati di dimensione dell’inserto breve e la rimozione del piccolo picco dei dati di dimensione dell’inserto grande. L’algoritmo di assemblaggio SOAPdenovo comprendeva tre passaggi principali. (1) Costruzione Contig: i dati della libreria di dimensioni brevi sono stati suddivisi in k-mer e costruiti utilizzando un grafico de Bruijn, che è stato semplificato rimuovendo le punte, unendo le bolle, rimuovendo la bassa copertura della connessione e rimuovendo piccole ripetizioni. Abbiamo ottenuto la sequenza contig collegando il percorso k-mer, risultando in un contig N50 2.104 bp e lunghezza totale 2.295.545.898 bp. (2) Costruzione dell’impalcatura: abbiamo ottenuto l ‘ 80% di tutte le letture accoppiate allineate riallineando tutte le letture utilizzabili sui contig. Quindi abbiamo calcolato la quantità di relazioni accoppiate-end condivise tra ogni coppia di contig, ponderato il tasso di coerentemente e in conflitto accoppiati-end, e poi costruito gli scaffold passo dopo passo. Di conseguenza, abbiamo ottenuto scaffold con un N50 7,168,038 bp e lunghezza totale 2,355,303,269 bp da estremità accoppiate di dimensioni ridotte, a estremità accoppiate distanti. (3) Gap closing: Per colmare le lacune all’interno degli scaffold costruiti, abbiamo utilizzato le informazioni accoppiate per recuperare le coppie di lettura per eseguire nuovamente un assemblaggio locale per queste letture raccolte. In sintesi, abbiamo chiuso l ‘87,7% delle lacune intra-scaffold, o l’ 85,8% della lunghezza del gap somma. La dimensione contig N50 è aumentata da 2.104 bp a 21.301 bp (Tabella 3). La dimensione dell’assemblea dell’impalcatura era 2.355.303.269 bp, che è vicina alla dimensione basata a assemblea del genoma di 2.374.716.107 bp riferita per il hyaena striato, Hyaena hyaena11 (adesione di NCBI: ASM300989v1). Abbiamo anche recuperato e annotato il genoma mitocondriale della iena maculata usando il programma mitoz16, che ha una lunghezza di 16.858 bp, simile ai primi genomi mitocondriali sequenziati per questa specie12.
La valutazione del progetto di genoma è stata eseguita esaminando la completezza degli ortologi a copia singola utilizzando BUSCO (versione 3.1.0)17, cercando nel database Mammaliaodb9 che contiene 4.104 gruppi di ortolog a copia singola. Un totale di 95.5% degli ortologhi è stato identificato come completo, 2.5% come frammentato e 2.0% come mancante, indicando un’alta qualità complessiva dell’assemblaggio del genoma della iena maculata. Dato che il 99,95% degli scaffold corti (<1k) ospitava solo 1.2% della lunghezza totale del genoma, abbiamo escluso questi scaffold per l’analisi a valle, tra cui elemento ripetitivo e gene caratteristica annotazione.
Annotazione degli elementi ripetitivi
Sia le ripetizioni tandem che gli elementi trasponibili (TE) sono stati ricercati e identificati attraverso il genoma di C. crocuta. Le ripetizioni tandem sono state identificate utilizzando Tandem Repeats Finder (TRF, v4.07) 18 e gli elementi trasposibili (TEs) sono stati identificati da una combinazione di approcci basati sull’omologia e de novo. Per la previsione basata sull’omologia, abbiamo usato RepeatMasker versione 4.0.619 con le impostazioni “- nolow-no_is-norna-engine ncbi “e RepeatProteinMask (un programma all’interno del pacchetto RepeatMasker) con le impostazioni” – engine ncbi-noLowSimple-pvalue 0.0001 ” per cercare TEs a livello di nucleotide e amminoacido sulla base di ripetizioni note (Fig. 3). RepeatMasker è stato applicato per l’identificazione a livello di DNA utilizzando una libreria personalizzata che ha combinato il Repbase21. 10 dataset20. A livello proteico, RepeatProteinMask è stato utilizzato per eseguire RMBlast contro il database proteico TE. Per ab initio prediction, RepeatModeler (v1.0. 8) 21 e LTR_FINDING (v1.06) 22 sono stati applicati per costruire la libreria di ripetizione de novo. Le sequenze di contaminazione e multi-copia nella libreria sono state rimosse e le sequenze rimanenti sono state classificate in base al risultato dell’ESPLOSIONE dopo l’allineamento alla banca dati SwissProt. Sulla base di questa libreria, abbiamo usato RepeatMasker per mascherare i TES omologhi e classificati (Fig. 4). Complessivamente, nella iena maculata sono stati identificati un totale di 826 Mb di elementi ripetitivi, comprendenti il 35,29% dell’intero genoma (Tabella 4).

Distribuzione del tasso di divergenza di ciascun tipo di elemento trasponibile (TE) nell’assemblaggio del genoma di Crocuta crocuta basato sulla predizione basata sull’omologia. Il tasso di divergenza è stato calcolato tra i TES identificati nel genoma utilizzando un metodo basato sull’omologia e la sequenza di consenso nella banca dati repbase20.

Distribuzione del tasso di divergenza di ciascun tipo di TE nell’assemblaggio del genoma di Crocuta crocuta basato sulla predizione ab initio. Il tasso di divergenza è stato calcolato tra le TES identificate nel genoma mediante predizione ab initio e la sequenza di consenso nella libreria TE prevista (vedi Metodi).
Annotazione genica codificante proteine
Abbiamo utilizzato ab initio prediction e approcci basati su omolog per annotare geni codificanti proteine e siti di splicing e isoforme di splicing alternative. Ab initio prediction è stata eseguita sul genoma a ripetizione mascherata utilizzando modelli genetici da umani, cane domestico, e gatto domestico utilizzando AUGUSTUS (versione 2.5.5)23, GENSCAN24, GlimmerHMM (versione 3.0.4)25, e SNAP (versione 2006-07-28) 26, rispettivamente. Un totale di 22.789 geni sono stati identificati con questo metodo. Le proteine omologhe di, Homo sapiens, Felis catus e Canis familiaris (dalla versione Ensembl 96) sono state mappate sul genoma della iena maculata usando tblastn (Blastall 2.2.26)27 con parametri “-e 1e-5”. Le sequenze allineate e le loro proteine di query sono state quindi sottoposte a GeneWise (versione 2.4.1) 28 per la ricerca di un allineamento giuntato accurato. Il set genetico finale (22.747) è stato raccolto unendo i risultati ab initio e homolog utilizzando una pipeline personalizzata (Tabella 5).
Annotazione della funzione genica
Le funzioni geniche sono state assegnate in base alla migliore corrispondenza ottenuta allineando le sequenze di codifica genica tradotte utilizzando BLASTP con i parametri “-e 1e-5” ai database SwissProt e TrEMBL (Uniprot release 2017-09). I motivi e i domini dei geni sono stati determinati da InterProScan (v5)29 rispetto ai database proteici tra cui ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 e PROSITE35. Gli ID di ontologia genica per ciascun gene sono stati ottenuti dalle corrispondenti voci SwissProt e TrEMBL. Tutti i geni sono stati allineati contro le proteine di KEGG e il percorso in cui il gene potrebbe essere coinvolto è stato derivato dai geni abbinati nel database di KEGG36. In sintesi, 22.166 (97,45%) dei geni codificanti proteine previsti sono stati annotati con successo da almeno uno dei sei database (Tabella 6).
famiglia del Gene costruzione e la ricostruzione della filogenesi
Per conoscere la storia filogenetica e l’evoluzione delle famiglie del gene di Crocuta crocuta, abbiamo gene cluster sequenze di sette specie (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) e l’Homo sapiens come l’outgroup (Ensembl release-96, Panthera leo da dati inediti) nel gene famiglie mediante orthoMCL (v2.0.9)37. I geni che codificano le proteine per le otto specie sono stati recuperati selezionando l’isoforma di trascrizione più lunga per ciascun gene per l’assegnazione a coppie a valle (costruzione del grafico). Abbiamo eseguito una ricerca BLASTP tutti contro tutti sulle sequenze proteiche di tutte le specie di riferimento, con un cut-off del valore E di 1e-5. Gene family construction ha impiegato l’algoritmo MCL38 con il parametro di inflazione di ‘1.5’. Un totale di 16.271 famiglie geniche di C. crocuta, H. sapiens, F. catus, A. melanoleuca sono state raggruppate. C’erano 11.671 famiglie di geni condivise da queste quattro specie, mentre 292 famiglie di geni contenenti 1.446 geni erano specifici di C. Crocuta (Fig. 5). Notevolmente, le famiglie geniche C. crocuta e F. catus condivise erano meno di C. crocuta e H. sapiens condivisi, il che potrebbe derivare da quel H. sapiens aveva un genoma e un’annotazione più completi.
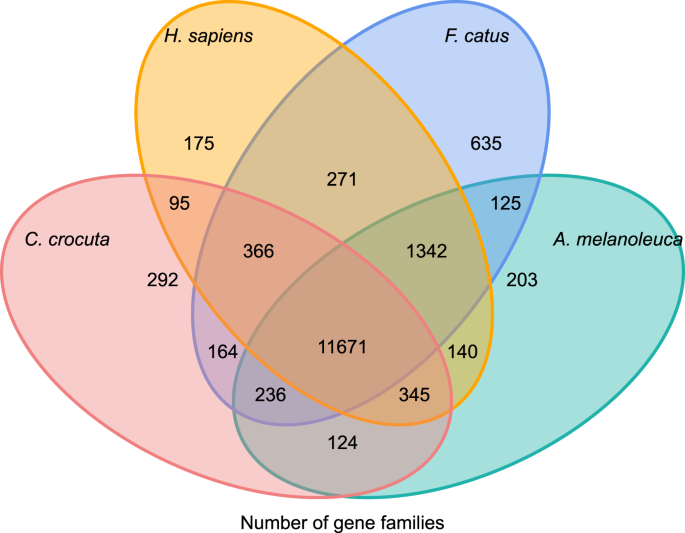
Diagramma di Venn che mostra il confronto di geni codificanti proteine condivisi e unici tra iena maculata, umano, gatto domestico e cane domestico basato sull’analisi ortologica.
Abbiamo identificato 6.601 geni ortologhi a copia singola per ricostruire l’albero filogenetico delle otto specie. Allineamenti di sequenze multiple di sequenze di amminoacidi per ciascun gene sono stati generati usando il MUSCOLO (versione 3.8.31)39 e tagliati usando Gblocks (0.91 b) 40, ottenendo regioni ben allineate con i parametri “-t = p-b3 = 8-b4 = 10-b5 = n-e = – st”. Abbiamo eseguito l’analisi filogenetica utilizzando il metodo di massima verosimiglianza implementato in PhyML (v3.0)41, utilizzando il modello JTT + G + I per la sostituzione degli aminoacidi (Fig. 6). La radice dell’albero è stata determinata riducendo al minimo l’altezza dell’intero albero tramite Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Infine, abbiamo stimato il tempo di divergenza tra le otto linee utilizzando MCMCTree dal pacchetto software PAML versione 4.442. Due priori basati sui reperti fossili sono stati utilizzati per calibrare il tasso di sostituzione, tra cui Boreoeutheria (91-102 MYA) e Carnivora (52-57 MYA)43. Coerentemente con studi precedenti, la iena maculata si raggruppa con le quattro specie incluse dai Felidae in un clade che definisce il sottordine Feliformia, che divergeva dai Caniformia (rappresentati dal cane domestico e dal panda gigante) 53.9 Mya44.

Albero filogenetico di C. crocuta e altre sette specie costruito con il metodo della massima verosimiglianza basato su 6.601 ortologhi a copia singola. Il tempo di divergenza è stato stimato utilizzando i due priori di calibrazione derivati dal database Time Tree (http://www.timetree.org), che sono contrassegnati da un rombo rosso. Tutti i tempi di divergenza stimati sono mostrati con intervalli di confidenza del 95% tra parentesi.