Spinal Muskelatrofi
Last ned Vår Faktaark For Spinal Muskelatrofi
Lær om MDAS COVID-19-respons
Hva er spinal muskelatrofi?
SPINAL muskelatrofi (SMA) er en genetisk sykdom som påvirker sentralnervesystemet, perifert nervesystem og frivillig muskelbevegelse (skjelettmuskulatur).
De fleste nervecellene som styrer musklene ligger i ryggmargen, som står for ordet spinal i navnet på sykdommen. SMA er muskuløs fordi den primære effekten er på muskler, som ikke mottar signaler fra disse nervecellene. Atrofi er den medisinske termen for å bli mindre, noe som vanligvis skjer med muskler når de ikke stimuleres av nerveceller.
SMA innebærer tap av nerveceller kalt motorneuroner i ryggmargen og er klassifisert som en motorneuronsykdom.
i den vanligste FORMEN FOR SMA (kromosom 5 SMA, ELLER SMN-relatert SMA), er det stor variasjon i alder, symptomer og progresjonshastighet. For å redegjøre for disse forskjellene, er kromosom 5-relatert SMA, som ofte er autosomal recessiv, klassifisert i type 1 til 4.
ALDEREN DER SMA-symptomer begynner, korrelerer grovt med i hvilken grad motorfunksjonen påvirkes: jo tidligere alder av utbruddet, desto større innvirkning på motorfunksjonen. Barn som viser symptomer ved fødselen eller i barndommen har vanligvis det laveste funksjonsnivået (type 1). SENERE OPPSTART SMA med mindre alvorlig kurs (type 2 og 3, og i tenåringer eller voksne, type 4) korrelerer generelt med stadig høyere nivåer av motorfunksjon.
For mer, se FORMER FOR SMA.
hva forårsaker SMA?
Kromosom 5 SMA er forårsaket av mangel på et motorneuronprotein kalt SMN, for » overlevelse av motorneuron.»Dette proteinet, som navnet antyder, synes å være nødvendig for normal motorneuronfunksjon. SMN spiller en sentral rolle i genuttrykk i motorneuroner. Dens mangel er forårsaket av genetiske feil (mutasjoner) på kromosom 5 i et gen som heter SMN1. Den vanligste mutasjonen I smn1-genet hos pasienter diagnostisert MED SMA er en sletting av et helt segment, kalt ekson 7.1 Nabo SMN2-gener kan delvis kompensere for ikke-funksjonelle SMN1-gener da det er 99% identitet mellom disse to genene.2
andre sjeldne FORMER FOR SMA (ikke-kromosom 5) er forårsaket av mutasjoner i andre gener ENN SMN1.3
For mer informasjon, inkludert detaljert lesing på sjeldne, ikke-kromosom 5-bundet SMA, se FORMER FOR SMA og Årsaker/Arv.
hva er SYMPTOMENE PÅ SMA?
SMA symptomer dekker et bredt spekter, alt fra mild til alvorlig.
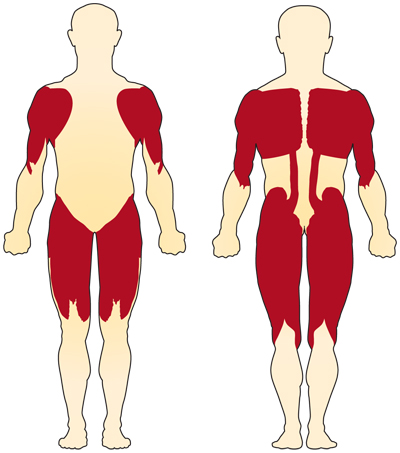
det primære symptomet på kromosom 5-relatert (SMN-relatert) SMA er svakhet i frivillige muskler. Musklene som er mest berørt er de som er nærmest midten av kroppen, for eksempel skuldre, hofter, lår og øvre del av ryggen. Nedre lemmer ser ut til å bli påvirket mer enn de øvre lemmer, og dype senereflekser reduseres.4
Spesielle komplikasjoner oppstår hvis musklene som brukes til å puste og svelge påvirkes, noe som resulterer i abnormiteter i disse funksjonene. Hvis musklene i ryggen svekkes, kan spinalkurvaturer utvikle seg.
det er stor variasjon i alder av utbrudd og nivå av motorfunksjon oppnådd i kromosom 5-relatert SMA. Disse er grovt korrelert med hvor mye funksjonelt SMN-protein som er tilstede i motorneuronene, som igjen korrelerer med hvor mange kopier AV SMN2-gener en person har. Sensorisk, mental og emosjonell funksjon er helt normalt i kromosom-5 SMA.
Noen FORMER FOR SMA er ikke knyttet til kromosom 5 eller SMN mangel. Disse skjemaene varierer sterkt i alvorlighetsgrad og i musklene som er mest berørt. Mens de fleste former, som kromosom 5-relatert form, påvirker det meste de proksimale musklene, finnes andre former som påvirker det meste distale muskler (de lenger unna kroppens sentrum) — i hvert fall i begynnelsen.
Se Tegn og Symptomer hvis Du vil ha Mer.
Hva er UTVIKLINGEN AV SMA?
i kromosom 5-relatert SMA, jo senere symptomene begynner og jo MER SMN protein det er, jo mildere sykdomsforløpet er sannsynlig å være. Mens i det siste, spedbarn med SMA vanligvis ikke overleve mer enn to år, i dag de fleste leger nå vurdere SMN-relaterte SMA å være et kontinuum og foretrekker ikke å gjøre stive spådommer om levealder eller svakhet basert strengt på alder av utbruddet.
SMA er den vanligste genetiske årsaken til dødelighet hos spedbarn.
hva er status for forskning PÅ SMA?
Forskning har fokusert på strategier for å øke kroppens produksjon AV SMN protein mangler i kromosom 5-relaterte former av sykdommen. Tilnærminger inkluderer metoder for å hjelpe motorneuroner overleve under ugunstige forhold.
På Desember. 23, 2016, US Food And Drug Administration (FDA) godkjente Spinraza (nusinersen) for behandling AV SMA. Spinraza er utviklet for å behandle den underliggende feilen I SMA, noe som betyr at det potensielt kan være effektivt å bremse, stoppe eller kanskje reversere symptomene på SMA. For Mer, se Spinraza Er Godkjent.
I Mai 2019 godkjente FDA Zolgensma (onasemnogene abeparvovac-xioi), den første gen-erstatningsterapien for en nevromuskulær sykdom. Zolgensma er en engangs intravenøs (i venen) infusjon for behandling av pediatriske pasienter yngre enn 2 år MED SMA med bi-alleliske mutasjoner I smn1-genet, inkludert de som er presymptomatiske ved diagnose. FOR mer informasjon les FDA Godkjenner AveXis ‘ Zolgensma For Behandling Av Spinal Muskelatrofi Hos Pediatriske Pasienter.
FOR Mer, se FORSKNING, SMA: Full Fart Fremover Og I Fokus: Spinal Muskelatrofi (SMA). For historier om familier som bor MED SMA, se VÅRE SMA stories På Strongly, THE mda blog.
I August 2020 godkjente FDA risdiplam (merkenavn Evrysdi*) for behandling av SMA hos voksne og barn to måneder eller eldre. Evysdi er en oral medisin designet for å øke nivåene AV SMN-proteinet ved å øke produksjonen FRA smn2″ backup » – genet.
- Ogino, S. & Wilson, Rb Genetisk testing Og risikovurdering for spinal muskelatrofi (SMA). Human Genetics (2002). doi: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Identifisering og karakterisering av et spinal muskelatrofi-bestemmende gen. Cell (1995). doi: 10.1016/0092-8674(95)90460-3
- Darras, B. T. Non-5q spinal muskulære atrofier: den alfanumeriske suppen tykner. Nevrologi (2011). doi: 10.1212 / WNL.0b013e3182267bd8
- Arnold, Wd, Kassar, D. & Kissel, Jt Spinal muskelatrofi: Diagnose og behandling i en ny terapeutisk epoke. Muskel Og Nerve (2015). doi: 10.1002 / mus.24497