- Colección de muestras, construcción de bibliotecas y secuenciación
- Control de calidad
- Estimación del tamaño del genoma
- Ensamblaje y evaluación del genoma
- Anotación de elementos repetitivos
- Anotación de genes codificadores de proteínas
- Anotación de función génica
- Construcción de familias de genes y reconstrucción de filogenia
Colección de muestras, construcción de bibliotecas y secuenciación
Se obtuvo ADN genómico de un espécimen macho de C. crocuta (NCBI taxonomy ID: 9678; Fig. 1) almacenado en el Zoológico Congelado ® en el Instituto de Investigación de Conservación del Zoológico de San Diego, EE. UU. (identificación del Zoológico congelado: KB4526).
El ADN genómico se extrajo con fenol-cloroformo seguido de purificación con precipitación de etanol 13. El ADN extraído fue analizado y visualizado en un 1.gel de agarosa al 5% en tampón de 1x TBE para verificar la presencia de ADN de alto peso molecular. La concentración y pureza del ADN se cuantificaron en un espectrofotómetro NanoDrop 2000 y un fluorómetro Qubit 2.0 (Thermo Fisher Scientific, EE.UU.) antes de enviarse a BGI-Shenzhen, China. Se obtuvo un total de 372 µg de ADN genómico, con una concentración de 0,418 µg/µL utilizando el Nanodrop 2000 y 0,245–0,399 µg/µL en base a cuatro lecturas replicadas utilizando el Fluorómetro Qubit 2.0. La relación de pureza de 260/280 fue de 1,95. Luego codificamos la muestra usando el gen del citocromo b (Cytb). Luego, de acuerdo con la estrategia de biblioteca de gradientes, construimos 13 bibliotecas de tamaño de inserción, con las siguientes longitudes de tamaño de inserción: 170 pb, 500 pb, 800 pb, 2 kbp, 5 kbp, 10 kbp, 20 kbp. Usamos el HiSeq. secuenciador 2000 (Illumina, EE. UU.) para secuenciar lecturas de extremo emparejado (PE) para cada biblioteca a través de 14 carriles. Se generó un total de aproximadamente 299 Gb de datos sin procesar de 13 bibliotecas, con lo que se alcanzó una profundidad de secuenciación (cobertura) de 149,25 (Tabla 1).
Control de calidad
Para minimizar los errores de montaje erróneo, filtramos las lecturas en bruto antes del ensamblaje del genoma de novo de acuerdo con los dos criterios siguientes. En primer lugar, se eliminaron las lecturas con más de 10 pb alineadas a la secuencia del adaptador (lo que permite una discrepancia de <= 3 pb). En segundo lugar, se descartaron lecturas con un 40% de bases con un valor de calidad inferior o igual a 10. Finalmente, se obtuvieron 190,4 G de datos con una cobertura de 95,2 (Tabla 2).
Estimación del tamaño del genoma
Se utilizaron tres bibliotecas de inserción corta (dos de 170 pb y una de 500 pb) para estimar el tamaño del genoma y la heterocigosidad de todo el genoma mediante análisis k-mer. Un total de aproximadamente 385 M de lectura de PE se sometieron a medidas14 para calcular la frecuencia k-mer. Luego, la distribución de k-mer fue ilustrada por Genomescope7 con parámetros «k = 17; longitud = 100; cobertura máxima = 1000». Se obtuvo un tamaño del genoma estimado de 2.003.681.234 pb, y heterocigosidad de 0,325% (Fig. 2).

17-estimación del tamaño del genoma. El eje x es profundidad (X), el eje y es la proporción que representa la frecuencia a esa profundidad dividida por la frecuencia total de todas las profundidades de cobertura. Sin considerar la tasa de error de secuencia, la tasa de heterocigosidad y la tasa de repetición del genoma, la distribución de 17 mer debería aproximarse a una distribución de Poisson.
Ensamblaje y evaluación del genoma
SOAPdenovo (V1.06) 15 se empleó para ensamblar el genoma de novo, siguiendo el filtrado de los datos de tamaño de inserción corto y eliminando el pico pequeño de los datos de tamaño de inserción grande. El algoritmo de ensamblaje de SOAPdenovo incluía tres pasos principales. (1) Construcción Contig: los datos de la biblioteca de tamaño de inserción corta se dividieron en k-mers y se construyeron utilizando un gráfico de Bruijn, que se simplificó eliminando puntas, fusionando burbujas, eliminando la baja cobertura de la conexión y eliminando pequeñas repeticiones. Se obtuvo la secuencia contig conectando la ruta k-mer, resultando en un contig N50 de 2.104 pb, y una longitud total de 2.295.545.898 pb. (2) Construcción de andamios: obtuvimos el 80% de todas las lecturas de extremos emparejados alineados realineando todas las lecturas utilizables en los contiguos. Luego calculamos la cantidad de relaciones de extremo emparejado compartidas entre cada par de contiguos, ponderamos la tasa de extremos emparejados consistentes y conflictivos, y luego construimos los andamios paso a paso. Como resultado, se obtuvieron andamios con un N50 de 7.168.038 pa, y una longitud total de 2.355.303.269 pa, desde extremos pareados de tamaño de inserción corto hasta extremos pareados de larga distancia. (3) Cierre de huecos: Para llenar los huecos dentro de los andamios construidos, utilizamos la información del extremo emparejado para recuperar los pares de lectura para hacer un ensamblaje local de nuevo para estas lecturas recopiladas. En resumen, cerramos el 87,7% de los huecos intra-andamio, o el 85,8% de la longitud del hueco de suma. El tamaño del contig N50 aumentó de 2.104 pb a 21.301 pb (Tabla 3). El tamaño del ensamblaje del andamio fue de 2.355.303.269 pb, que se aproxima al tamaño del genoma basado en el ensamblaje de 2.374.716.107 pb reportado para la hyaena rayada, Hyaena hyaena11 (acceso NCBI: ASM300989v1). También recuperamos y anotamos el genoma mitocondrial de la hiena manchada utilizando el programa mitoz16, que tiene una longitud de 16.858 pb, similar a los primeros genomas mitocondriales secuenciados para esta especia12.
La evaluación del borrador del genoma se realizó observando la integridad de los ortólogos de copia única utilizando BUSCO (versión 3.1.0)17, buscando en la base de datos Mammaliaodb9, que contiene 4.104 grupos de ortólogos de copia única. Un total del 95,5% de los ortólogos se identificaron como completos, el 2,5% como fragmentados y el 2,0% como faltantes, lo que indica una alta calidad general del conjunto del genoma de la hiena manchada. Dado que el 99,95% de los andamios cortos (<1k) albergaban solo 1.2% de la longitud total del genoma, excluimos estos andamios para el análisis posterior, incluida la anotación de elementos repetitivos y características genéticas.
Anotación de elementos repetitivos
Se buscaron e identificaron repeticiones en tándem y elementos transponibles (TE) en todo el genoma de C. crocuta. Se identificaron repeticiones en tándem utilizando el Buscador de repeticiones en Tándem (TRF, v4. 07) 18 y se identificaron elementos transponibles (TEs) mediante una combinación de enfoques basados en homología y de novo. Para la predicción basada en homología, utilizamos la versión 4.0 de RepeatMasker.619 con la configuración»- nolow-no_is-norna-engine ncbi «y RepeatProteinMask (un programa dentro del paquete RepeatMasker) con la configuración»- engine ncbi-noLowSimple-pvalue 0.0001 » para buscar TEs a nivel de nucleótidos y aminoácidos en función de repeticiones conocidas (Fig. 3). Se aplicó RepeatMasker para la identificación a nivel de ADN utilizando una biblioteca personalizada que combinaba el conjunto de datos Repbase21.1020. A nivel de proteína, se utilizó una máscara de repetición para realizar un RMBBLAST contra la base de datos de proteínas TE. Para la predicción ab initio, RepeatModeler (v1.0.8)21 y LTR_FINDING (v1.06) se aplicaron 22 para construir la biblioteca de repetición de novo. Se eliminaron las secuencias de contaminación y copias múltiples de la biblioteca y las secuencias restantes se clasificaron de acuerdo con el resultado de la EXPLOSIÓN tras la alineación con la base de datos SwissProt. En base a esta biblioteca, utilizamos RepeatMasker para enmascarar las TEs homólogas y clasificarlas (Fig. 4). En total, se identificaron 826 Mb de elementos repetitivos en la hiena manchada, que representan el 35,29% de todo el genoma (Tabla 4).

Distribución de la tasa de divergencia de cada tipo de elemento transponible (TE) en el ensamblaje del genoma de Crocuta crocuta basada en la predicción basada en la homología. La tasa de divergencia se calculó entre las EEt identificadas en el genoma utilizando un método basado en homología y la secuencia de consenso en la base de datos Repbase 20.

Distribución de la tasa de divergencia de cada tipo de TE en el ensamblaje del genoma de Crocuta crocuta basada en la predicción ab initio. La tasa de divergencia se calculó entre las EEt identificadas en el genoma mediante predicción ab initio y la secuencia de consenso en la biblioteca de EET predicha (ver Métodos).
Anotación de genes codificadores de proteínas
Utilizamos métodos de predicción ab initio y basados en homólogos para anotar genes codificadores de proteínas, así como sitios de empalme e isoformas de empalme alternativas. La predicción Ab initio se realizó en el genoma enmascarado repetido utilizando modelos genéticos de humanos, perros domésticos y gatos domésticos utilizando AUGUSTUS (versión 2.5.5)23, GENSCAN24, GlimmerHMM (versión 3.0.4)25 y SNAP (versión 2006-07-28)26, respectivamente. Un total de 22,789 genes fueron identificados por este método. Las proteínas homólogas de Homo sapiens, Felis catus y Canis familiaris (de la liberación Ensembl 96) se asignaron al genoma de la hiena manchada utilizando tblastn (Blastall 2.2.26)27 con parámetros «-e 1e-5». Las secuencias alineadas, así como sus proteínas de consulta, se enviaron a GeneWise (versión 2.4.1)28 para buscar una alineación empalmada precisa. El conjunto final de genes (22.747) se recopiló mediante la fusión de resultados basados en ab initio y homólogos utilizando una canalización personalizada (Tabla 5).
Anotación de función génica
Las funciones génicas se asignaron de acuerdo con la mejor coincidencia obtenida al alinear secuencias de codificación génica traducidas utilizando BLASTP con parámetros «-e 1e-5» a las bases de datos SwissProt y TrEMBL (versión de Uniprot 2017-09). Los motivos y dominios de los genes se determinaron mediante InterProScan (v5) 29 contra bases de datos de proteínas como ProDom30, PRINTS31, Pfam32, SMART33, PANTHER34 y PROSITE35. Los identificadores de ontología génica de cada gen se obtuvieron de las entradas SwissProt y TrEMBL correspondientes. Todos los genes estaban alineados contra las proteínas KEGG, y la vía en la que el gen podría estar involucrado se derivó de los genes coincidentes en la base de datos KEGG36. En resumen, 22.166 (97,45%) de los genes codificadores de proteínas previstos fueron anotados con éxito por al menos una de las seis bases de datos (Tabla 6).
Construcción de familias de genes y reconstrucción de filogenia
Para obtener información sobre la historia filogenética y la evolución de familias de genes de Crocuta crocuta, agrupamos secuencias de genes de siete especies (Felis catus, Canis familiaris, Ailuropoda melanoleuca, Crocuta crocuta, Panthera pardus, Panthera leo, Panthera tigris altaica) y Homo sapiens como grupo externo (Ensembl release-96, Panthera leo a partir de datos no publicados) en familias de genes utilizando orthoMCL (v2.0.9)37. Los genes codificadores de proteínas para las ocho especies se recuperaron seleccionando la isoforma de transcripción más larga para cada gen para la asignación por pares aguas abajo (construcción de gráficos). Realizamos una búsqueda de BLASTP todo contra todo en las secuencias de proteínas de todas las especies de referencia, con un valor de corte E de 1e-5. La construcción de la familia de genes empleó el algoritmo MCL38 con el parámetro de inflación de ‘1,5’. Se agruparon un total de 16.271 familias de genes de C. crocuta, H. sapiens, F. catus y A. melanoleuca. Había 11.671 familias de genes compartidas por estas cuatro especies, mientras que 292 familias de genes que contenían 1.446 genes eran específicos de C. Crocuta (Fig. 5). Notablemente, las familias de genes compartidas por C. crocuta y F. catus fueron menores que las compartidas por C. crocuta y H. sapiens, lo que podría resultar de que H. sapiens tuviera un genoma y anotación más completos.
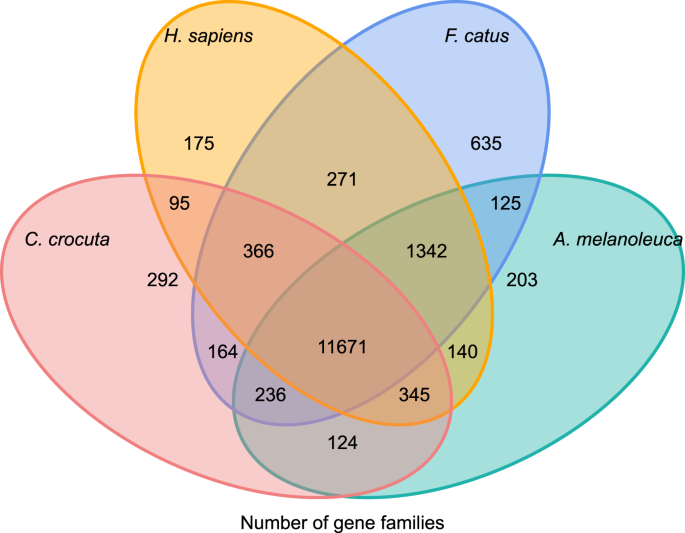
Diagrama de Venn que muestra la comparación de genes codificadores de proteínas compartidos y únicos entre hienas manchadas, humanos, gatos domésticos y perros domésticos según el análisis ortológico.
Se identificaron 6.601 genes ortólogos de una sola copia para reconstruir el árbol filogenético de las ocho especies. Se generaron alineaciones múltiples de secuencias de aminoácidos para cada gen utilizando MÚSCULO (versión 3.8.31)39, y se recortaron utilizando bloques G (0.91 b)40, logrando regiones bien alineadas con los parámetros «-t = p-b3 = 8-b4 = 10-b5 = n-e = – st». Se realizó un análisis filogenético utilizando el método de máxima verosimilitud implementado en PhyML (v3.0)41, utilizando el modelo JTT + G + I para la sustitución de aminoácidos (Fig. 6). La raíz del árbol se determinó minimizando la altura de todo el árbol a través de Treebest (v1.9.2; http://treesoft.sourceforge.net/treebest.shtml). Finalmente, se estimó el tiempo de divergencia entre los ocho linajes utilizando MCMCTree del paquete de software PAML versión 4.442. Se utilizaron dos antecedentes basados en el registro fósil para calibrar la tasa de sustitución, incluidos Boreoeutheria (91-102 MA) y Carnivora (52-57 MA)43. De acuerdo con estudios anteriores, las hienas manchadas se agrupan con las cuatro especies incluidas de los Felidae en un clado que define el suborden Feliformia, que diverge del Caniformia (representado por el perro doméstico y el panda gigante) 53,9 Ma44.

Árbol filogenético de C. crocuta y otras siete especies construido por el método de máxima verosimilitud basado en 6.601 ortólogos de copia única. El tiempo de divergencia se estimó utilizando los dos antecedentes de calibración derivados de la base de datos del Árbol del Tiempo (http://www.timetree.org), que están marcados con un rombo rojo. Todos los tiempos de divergencia estimados se muestran con intervalos de confianza del 95% entre paréntesis.