„być albo nie być: oto jest pytanie.”Podczas gdy wszyscy jesteśmy gotowi na decyzje o życiu lub śmierci, ta egzystencjalna dychotomia jest wyjątkowo poważna dla komórek embrionalnych. Programowana śmierć komórki, zwana apoptozą, * jest normalną częścią rozwoju. W nicieniu C. elegans, w którym możemy policzyć liczbę komórek, dokładnie 131 komórek umiera zgodnie z normalnym wzorcem rozwojowym. Wszystkie komórki tego nicienia są „zaprogramowane” na śmierć, chyba że zostaną aktywnie poinformowane, aby nie poddawały się apoptozie. U ludzi, aż 1011 komórek umiera w każdym dorosłym każdego dnia i są zastępowane przez inne komórki. (Rzeczywiście, masa komórek, które tracimy każdego roku w wyniku normalnej śmierci komórkowej, jest zbliżona do całej masy naszego ciała!) Wewnątrz macicy nieustannie tworzyliśmy i niszczyliśmy komórki, i wygenerowaliśmy około trzy razy więcej neuronów niż w końcu, kiedy się urodziliśmy. Lewis Thomas (1992) trafnie zauważył,
zanim się urodziłem, więcej mnie umarło niż przeżyło. Nic dziwnego, że nie pamiętam; w tym czasie przez dziewięć miesięcy przechodziłem przez mózg po mózgu, w końcu wymyślając jedyny model, który mógłby być ludzki, wyposażony w język.
apoptoza jest niezbędna nie tylko do prawidłowego rozmieszczenia i orientacji neuronów, ale także do generowania przestrzeni ucha środkowego, otworu pochwy i przestrzeni między palcami u rąk i nóg Saunders and Fallon 1966roberts and Miller 1998; Rodriguez et al. 1997. Apoptoza ściera niepotrzebne struktury, kontroluje liczbę komórek w poszczególnych tkankach i rzeźbi złożone narządy.
różne tkanki używają różnych sygnałów do apoptozy. Jednym z sygnałów często stosowanych u kręgowców jest białko morfogenetyczne Kości 4 (BMP4). Niektóre tkanki, takie jak tkanka łączna, reagują na BMP4 poprzez różnicowanie w kości. Inne, takie jak ektoderma żaby gastrula, reagują na BMP4 różnicując się w skórę. Jeszcze inne, takie jak komórki grzebienia nerwowego i zawiązki zębów, reagują degradując swoje DNA i obumierając. Na przykład w rozwijającym się zębie liczne czynniki wzrostu i różnicowania są wydzielane przez węzeł szkliwa. Po wyrośnięciu guzka węzeł szkliwa syntetyzuje BMP4 i zamyka się przez apoptozę (patrz rozdział 13; Vaahtokari et al. 1996b).
w innych tkankach komórki są” zaprogramowane „na śmierć i pozostaną żywe tylko wtedy, gdy obecny jest jakiś czynnik wzrostu lub różnicowania, aby je” uratować”. Dzieje się tak podczas rozwoju czerwonych krwinek ssaków. Prekursory krwinek czerwonych w wątrobie myszy potrzebują hormonu erytropoetyny, aby przetrwać. Jeśli go nie otrzymają, przechodzą apoptozę. Receptor erytropoetyny działa poprzez szlak jak-STAT, aktywując czynnik transkrypcyjny Stat5. W ten sposób ilość obecnej erytropoetyny może określić, ile czerwonych krwinek dostaje się do krążenia.
jedna ze ścieżek apoptozy została w dużej mierze wytyczona poprzez badania genetyczne C. elegans. Stwierdzono, że białka kodowane przez geny ced-3 i CED-4 były niezbędne do apoptozy, ale w komórkach, które nie przeszły apoptozy, geny te zostały wyłączone przez produkt genu ced-9 (rysunek 6.27 a; Hengartner i wsp . 1992). Białko CED – 4 jest czynnikiem aktywującym proteazę, który aktywuje CED-3, proteazę, która inicjuje zniszczenie komórki. Mutacje, które inaktywują białko CED-9 powodują liczne komórki, które normalnie przetrwałyby, aby aktywować ich geny ced-3 i ced-4 i umrzeć. Prowadzi to do śmierci całego zarodka. Z drugiej strony, mutacje CED-9 powodują powstawanie białka CED-9 w komórkach, które w przeciwnym razie obumarłyby. Tak więc Gen ced-9 wydaje się być binarnym przełącznikiem, który reguluje wybór między życiem a śmiercią na poziomie komórkowym. Możliwe jest, że każda komórka w zarodku nicienia jest gotowa do śmierci, a komórki, które przeżyją, są uratowane przez aktywację genu ced-9.
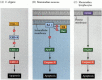
rysunek 6.27
ścieżki apoptozy u nicieni i ssaków. (A) w C. elegans białko CED-4 jest czynnikiem aktywującym proteazę, który może aktywować proteazę CED-3. Proteaza CED-3 inicjuje procesy niszczenia komórek. CED-9 może hamować CED-4 (i CED-9 może być (więcej…)
białka CED-3 i CED-4 tworzą centrum szlaku apoptozy, które jest wspólne dla wszystkich badanych zwierząt. Wyzwalaczem apoptozy może być sygnał rozwojowy, taki jak konkretna cząsteczka (taka jak BMP4 lub glikokortykosteroidy) lub utrata przyczepności do matrycy. Każdy typ cue może aktywować białka CED-3 lub CED-4 lub inaktywować cząsteczki CED-9. U ssaków homologi białka CED-9 należą do rodziny genów Bcl-2. Rodzina ta obejmuje Bcl-2, Bcl-X i podobne geny. Funkcjonalne podobieństwa są tak silne, że jeśli aktywny ludzki gen BCL-2 zostanie umieszczony w embrionach C. elegans, zapobiega to normalnie występującym zgonom komórek w embrionach nicieni(Vaux et al. 1992). W rozwoju czerwonych krwinek kręgowców (wspomnianym powyżej) czynnik transkrypcyjny Stat5 aktywowany przez erytropoetynę działa poprzez wiązanie się z promotorem genu Bcl-X, gdzie aktywuje syntezę tego białka przeciw apoptozie (Socolovsky i wsp. 1999).
mammalian homologue of CED-4 is called Apaf-1 (apoptotic protease activating factor-1) i bierze udział w zależnej od cytochromu aktywacji ssaczych homologów CED-3, proteaz caspase-9 i caspase-3 (Shaham and Horvitz 1996; Cecconi et al. 1998; Yoshida et al. 1998). Aktywacja kaspaz powoduje autodigitację komórki. Kaspazy są silnymi proteazami i trawią komórki od wewnątrz. Białka komórkowe są rozszczepiane, a DNA jest fragmentowane.†
podczas gdy nicienie z niedoborem apoptozy z niedoborem CED-4 są żywotne (pomimo posiadania 15% więcej komórek niż robaki dzikiego typu), myszy z mutacjami utraty funkcji dla kaspazy-3 lub kaspazy-9 umierają około urodzenia z masywnego przerostu komórek w układzie nerwowym (rysunek 6.28; Kuida i in. 1996, 1998; Jacobson et al. 1997). Myszy homozygotyczne dla ukierunkowanych delecji Apaf-1 mają poważne nieprawidłowości czaszkowo-twarzowe, przerost mózgu i wstęgę między palcami.
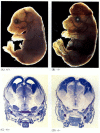
rysunek 6.28
zakłócenie normalnego rozwoju mózgu poprzez blokowanie apoptozy. U myszy, u których wyeliminowano kaspazę-9 lub Apaf-1, nie występuje prawidłowa apoptoza nerwowa. U myszy z niedoborem kaspazy 9 nadproliferacja neuronów mózgu jest oczywista na poziomie morfologicznym (więcej …)
u ssaków istnieje więcej niż jedna droga do apoptozy. Na przykład na apoptozę limfocytów nie ma wpływu delecja Apaf-1 lub kaspazy-9 i działa na oddzielny szlak zapoczątkowany przez białko CD95 (Fig. 6.27B, C) różne kaspazy mogą funkcjonować w różnych typach komórek w celu pośredniczenia w Sygnałach apoptotycznych (Hakem et al. 1998; Kuida et al. 1998).
strona internetowa
6.7 zastosowania apoptozy. Apoptoza jest wykorzystywana w wielu procesach w trakcie rozwoju. Ta strona internetowa bada rolę apoptozy w takich zjawiskach, jak rozwój komórek zarodkowych Drosophila i oczy ślepych ryb jaskiniowych. http://www.devbio.com/chap06/link0607.shtml
Box
interakcje komórka-komórka i szansa w określaniu typów komórek.
Przypisy
*
apoptoza (oba „p”S są wymawiane) pochodzi od greckiego słowa oznaczającego naturalne procesy liści spadających z drzew lub płatków z kwiatów. Jest aktywny i może być ewolucyjnie wybierany. Inny rodzaj śmierci komórki, martwica, jest patologiczną śmiercią spowodowaną czynnikami zewnętrznymi, takimi jak zapalenie lub toksyczne uszkodzenie.
†
istnieją pewne dowody (Patrz Barinaga 1998a, b; Saudou et al. 1998), że aktywacja szlaku apoptozy w dorosłych neuronach może być odpowiedzialna za patologię choroby Alzheimera i udaru mózgu. Fragmentacja DNA jest jednym z głównych sposobów rozpoznawania apoptozy i jest obserwowana w regionach mózgu dotkniętych tymi chorobami. Jego fragmentacja na kawałki o określonej wielkości(chronione i trzymane razem przez nukleosomy) może być spowodowana trawieniem polimerazy Poli (ADP-rybozy) (PARP) przez kaspazę-3 (Lazebnik i in. 1994). PARP rozpoznaje i naprawia pęknięcia DNA. Więcej informacji na temat innych ścieżek prowadzących do apoptozy znajduje się w Green 1998.